 | |||||||||||||||
 |
 |
 |
|
 |
|
 |
|
 |
|
 |
|
 |
|
 |
 |
AMBER Archive (2009)
Subject: [AMBER] Can we use AMBER?
From: Casey,Richard (Richard.Casey_at_ColoState.EDU)
Date: Thu Jan 08 2009 - 11:25:57 CST
- Next message: Beale, John: "[AMBER] lone pairs on CYX and MET"
- Previous message: wl2290_at_columbia.edu: "[AMBER] mairun"
- Next in thread: Ross Walker: "RE: [AMBER] Can we use AMBER?"
- Reply: Ross Walker: "RE: [AMBER] Can we use AMBER?"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
Hello,
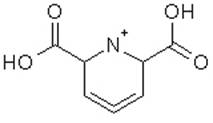
We would like to run molecular dynamics simulations for the compound shown below. Is Amber the correct force field for this? Or are there other more appropriate force fields?
[cid:image001.jpg_at_01C9717B.7E957550]
--------------------------------------------
Richard
_______________________________________________
AMBER mailing list
AMBER_at_ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
- Next message: Beale, John: "[AMBER] lone pairs on CYX and MET"
- Previous message: wl2290_at_columbia.edu: "[AMBER] mairun"
- Next in thread: Ross Walker: "RE: [AMBER] Can we use AMBER?"
- Reply: Ross Walker: "RE: [AMBER] Can we use AMBER?"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
|
VU Home |
VUMC Home |
People Finder |
University Calendar
webmaster- modified on January 30, 2009 |