Structure and
Function of Circadian Clock Proteins
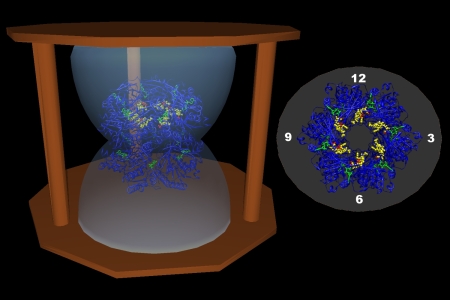
Circadian
clocks are self-sustained biochemical
oscillators. Their properties include temperature compensation, a time
constant of approximately 24 h, and high precision. Recent research has shown that the KaiABC
circadian clock from the cyanobacterium S. elongatus can be
reconstituted in vitro from the three proteins KaiC, KaiA and KaiB in
the presence of ATP. This renders the KaiABC molecular timer a unique
target of biochemical and biophysical studies. We are characterizing
this clock system using X-ray crystallography in combination with
electron microscopy and mutagenetic studies. We have recently
determined the crystal
structure of the KaiC auto-kinase and auto-phosphatase and have
presented three-dimensional models of binary KaiA-KaiC and KaiB-KaiC
complexes that shed light on the roles of the KaiA and KaiB proteins in
controlling the KaiC phosphorylation status. Collaborations with
the labs of Carl
H. Johnson and
Phoebe L.
Stewart at Vanderbilt University.
A flurry of crystallographic and NMR
studies have recently led to the
structural characterization of the cyanobacterial KaiA, KaiB and KaiC
proteins (reviewed in c.v. ref. 92 abd ref. 119 and highlighted by A.
Yarnell in
C&EN)
http://pubs.acs.org/isubscribe/journals/cen/82/i31/html/8231sci2.html
See also the discussion of circadian clock protein structures in the PDB 'molecule of the month' series (Jan, 2008):
http://pdb101.rcsb.org/motm/97
For more information on circadian clock
advances related to
the KaiABC family of proteins please go to the following link at
Vanderbilt's Exploration science e-mag Page:
https://www.mc.vanderbilt.edu/reporter/index.html?ID=3520
Supported by grant NIH R01 GM073845 and GM081646
|
 |
Microfluidic Integrated Transduction RealNose and Olfactory Receptors
Animal noses have evolved to rapidly detect small airborne and soluble molecules at minute concentrations. The range of odorants detected is chemically diverse and seemingly infinite. The sensitivity of the animal nose is exemplified in the canine, with over 1000 functional olfactory receptor (OR) genes that allow detection of many compounds at the level of parts per trillion (ppt). Despite decades of efforts, artificial noses are still embarrassingly inferior to their natural counterparts. The key to creating sensitive real-time olfactory sensors is to study and utilize the corner stone of mammalian scent detection -- the olfactory receptor. Our laboratory is part of a research team centered at MIT that studies ORs and aims to develop OR-based (RealNose) sensors. The currently available structures of ORs are rough estimations based on computational modeling and comparisons to bovine rhodopsin. No detailed molecular structure for any OR has yet been determined by X-ray diffraction. An accurate 3-dimensional OR model will likely provide insights into the mechanism by which organic compounds (odors) activate the olfactory system. Therefore, in addition to the development of the RealNose sensor, a central focus of the project is the expression and crystallization of OR membrane proteins suitable for X-ray crystal structure determination.
Supported by DARPA contract HR0011-09-C-0012
|
 |
|
Protein-Nucleic
Acid Interactions
We have
embarked on an
investigation of the structural basis that underlies the recognition of
RNA-DNA hybrids by E. coli RNase HI. The enzyme binds to the hybrid
duplex and degrades the RNA strand. Although the X-ray crystal
structure of RNase HI from E. coli was reported a decade ago, there is
presently no crystal structure of the enzyme-substrate complex.
Although we (c.v. refs. 18 and 22) and others have studied the
structures of chimeric RNA-DNA molecules by X-ray crystallography,
neither the structures of substrates and the enzyme alone nor those of
modeled complexes have yielded a satisfactory explanation for the
substrate specificity of the enzyme (c.v. ref. 63). RNase H is a key
player in replication and transcription (reverse transcriptases also
feature an RNase H domain) and is therefore of fundamental biological
importance. This project is a logical extension of our previous work on
nucleic acid structure and antisense oligonucleotide design. RNase H is
believed to play an important role in the suppression of a particular
message by an antisense oligonucleotide (another possible mode of
action of an antisense oligonucleotide is via a steric block
mechanism). Obviously, the availability of the three-dimensional
structure of a complex between RNase H and its substrate would be very
useful for the design of nucleic acid modifications that allow
recruitment of the enzyme to the site of hybridization and subsequent
RNA cleavage. In the absence of such a structure, a correlation of the
conformations in a duplex environment (DNA, RNA-DNA hybrid) of
nucleotide analogs with the susceptibilities to cleavage by RNase H of
their hybrids with RNA should yield valuable insights regarding the
features of the hybrid duplex that underlie enzyme
substrate-recognition and processivity. Nucleotide analogs that are
being analyzed in this manner in our laboratory include arabino nucleic
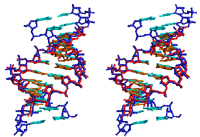
acid (ANA), 2'-F-ANA (see the structure of the duplex between 2'-F-ANA
and RNA depicted above) and a host of 2'-O-modified ribonucleotide
analogs.
|
 |
|
Structure Assisted Approach to the Discovery
of New Therapies for Neurodegenerative Diseases
Death-associated
protein kinase (DAPK) is the first described member of
a novel family of pro-apoptotic and tumor-suppressive serine/threonine
kinases. In collaboration with the laboratory of D. Martin Watterson at
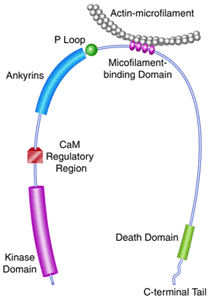
Northwestern University, we determined the crystal structure of the
catalytic domain of DAPK (c.v. refs. 74; a schematic of the domain
structure of DAPK is depicted above). The structures studied include
apo-form, the complex with AMPPnP as well as a ternary complex
consisting of kinase, AMPPnP and either Mg2+ or Mn2+.
A comparison between these structures of DAPK and nucleotide
triphosphate complexes of several other kinases revealed several unique
features of the DAPK catalytic domain. For example, a highly ordered
basic loop in the N-terminal domain may be of importance in enzyme
regulation.
In parallel with the structural work DAPK's
preferences for
phosphorylation site sequences was determined using a positional
scanning peptide substrate library (c.v. ref. 75). An enzyme assay for
DAPK was developed and then used to measure activity in adult brain as
well as to monitor protein purification based on the chemical and
physical properties of the DAPK cDNA open reading frame. The results of
the two studies allowed insight into DAPK's substrate preferences and
regulation and provide a foundation for proteomic investigations and
inhibitor discovery.
Current inhibitor discovery efforts using a
structure-assisted approach
have led to the identification of a small molecule lead with high
affinity and specificity for DAPK that attenuates hypoxia-ischemia
induced brain damage in vivo (c.v. ref. 89).
For more information on DAPK please follow
this link to Vanderbilt's Exploration science e-mag page:
http://www.vanderbilt.edu/exploration/home_egli.htm
|
 |
|
Structural Genomics
A first
phase of the
Human Genome Project has recently been completed and has produced a
working map of the entire human genome. Knowledge of its DNA sequence,
which is estimated to code for (surprisingly) only ca. 30,000 proteins,
is necessary but not sufficient for a complete understanding of human
biology, or that of other living systems. The next logical step is to
determine the biochemical functions and structures of these proteins. A
long-range goal is to determine the structures of all 30,000 proteins.
This is a formidable task and is unobtainable in the near term.
My interest in the type of research now
commonly termed structural
genomics lies not in the development of high-throughput crystallization
or structure determination. This is best left to companies and
government laboratories. However, one of the main goals of the centers
currently supported by the National Institutes of Health is the
production of as many new structures as possible within the next five
years. Thus, the projects will leave little time and money for further
work aimed at linking emerging structures to biological function. This
task is a suitable one for academic research. We are using
state-of-the-art sequence analysis and X-ray crystallographic methods
combined with functional assays to determine function from structure
for a handful of highly conserved genes of presently unknown function.
These include the YrdC protein from E. coli and the Maf protein from B.
subtilis for which crystal structures have recently been determined in
my laboratory (c.v. refs. 69 and 64, resp.). The above illustration
depicts the crystal structure of Maf along with a sequence alignment
for the maf-family of genes.
|
 |
|
Structure-Based Design of Antisense and Ribozyme
Therapeutics / Nucleic Acid Etiology
Chemically
modified
oligonucleotides are currently being investigated as antisense and
antigene reagents with potential therapeutic applications. Interference
with biological information transfer can occur at a variety of stages.
Thus, targeting either mRNA synthesis (transcription - antigene
approach) or protein synthesis (translation - antisense approach) may
allow a modulation of gene expression. The great potential of the
antisense strategy consists in the high specificity of hybridization
between antisense strand and messenger RNA via formation of
Watson-Crick base pairs, offering the opportunity of rationally
designing nucleic acid drugs. In 1998, the US Food and Drug
Administration (FDA) approved the first antisense drug, Vitravene™
(Formivirsen), a DNA phosphorothioate oligonucleotide against
cytomegalovirus-induced retinitis in AIDS patients.
We are pursuing a
structure-based approach to define the principles that underlie the
thermal stability (c.v. refs. 76, 57, 50, 45) and nuclease resistance
of oligonucleotides and how chemical modifications affect these
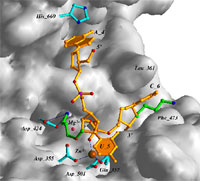
properties. The above illustration shows the detailed interactions of
an oligodeoxynucleotide containing 2'-O-(3-aminopropyl)-modified
nucleotides at the active site of the 3'-5'-exonuclease from DNA Pol I
Klenow fragment based on a crystal structure of the complex (c.v. ref.
61). My laboratory has published more structures of oligonucleotide
analogs than any other research group worldwide and we will continue
these efforts, focusing on modifications that are of potential
therapeutic interest as well as on those studied in the context of
nucleic acid etiology. Efforts regarding the latter are currently
concentrated on the crystal structure determination of a
2',3'-dideoxyglucopyranose nucleic acid duplex (homo-DNA) and on the
origins of the established cross-pairing between TNA (tetrose nucleic
acid) and both DNA and RNA (c.v. ref. 82).
|
 |
|
RNA Structure
In many
viruses,
including tumor- and retro-viruses, the programmed -1 ribosomal
frameshifting of polycistronic mRNA regulates the relative level of
structural and enzymatic proteins important for efficient viral
assembly. The -1 shift in reading frames causes stop codon readthrough,
and results in production of a single fusion protein. For example, in
the Rous sarcoma retrovirus, the pol gene that encodes integrase,
protease and reverse transcriptase is expressed with the upstream gag
gene (encoding virus core proteins) through a gag-pol fusion protein.
The mature products are later obtained by processing the poly-protein
precursor. The -1 frameshifting is not only found in retroviruses but
also in coronaviruses, yeast and plant viruses as well as bacterial
systems. Frameshifting levels can range from 1 to over 30% in different
systems to produce gene products in a functionally appropriate ratio.
However, the mechanism of ribosomal frameshifting is not understood. It
is postulated that a complex mRNA structure 6-8 nucleotides downstream
from the "slippery sequence", in many cases a pseudoknot, leads to
ribosomal pausing and the simultaneous slippage of both aminoacyl and
peptidyl tRNAs toward the 5'-direction by one base.
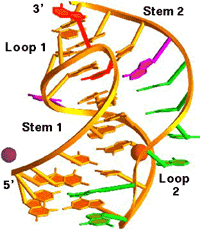
We participated in the
determination of the 1.6 Å resolution crystal structure of a
28-nucleotide pseudoknot from Beet Western Yellow Virus (BWYV, see
illustration above; c.v. ref. 53). In the meantime, we have
refined this structure to 1.25 Å and we have determined the
structure
of a second crystal form to 2.85 Å resolution (c.v. ref.
78).
The next phase of this project involves the correlation of the mutation
data collected in the laboratory of Dr. Alexander Rich at MIT with the
structures of mutated RNA pseudoknots. This will entail the
crystallization and structure determination of pseudoknots with
sequence altertations that cause drastic changes in the frameshifting
activity. In the more distant future it may be feasible to study the
interactions of a viral message that contains a pseudoknot with the
ribosomal proteins at the entry site in crystals of the E. coli
ribosome.
|
 |
|
DNA Crystallography
The
interactions
between double helical DNA and cations, specifically mono- and divalent
metal ions have recently received increased attention. Molecular
Dynamics simulations, solution NMR and X-ray crystallography have all
shed light on the coordination of ions in the major and minor grooves
of DNA. Metal ion interactions may play key roles in the control of DNA
conformation and topology, but despite progress in locating the ions
and determining their precise binding modes, it remains difficult to
figure out just how important ions really are (c.v. ref. 79).
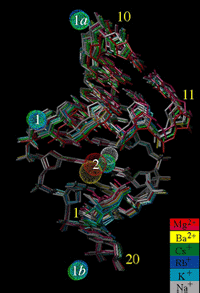
Most of the crystallographic investigations of DNA-ion coordination, in
particular those concerning potential binding of alkali and earth
alkali metal ions to so-called A-tracts, focused on the Dickerson-Drew
dodecamer. We would like to expand our previous investigations (c.v.
refs.
52, 56, 58) on other sequences containing longer A-tracts. We have
demonstrated the usefulness of the single-wavelength anomalous
dispersion (SAD) technique for locating alkali metal ions in DNA
crystals (see the above illustration, depicting ion coordination to an
A-form DNA duplex) and for determining the structures of the latter (c.v.
ref. 70).
|
 |
|
Phasing Strategies
For
proteins and
enzymes, selenium has proven to be an effective anomalous scattering
center that can be readily introduced into recombinant proteins in the
form of selenomethionine. Selenomethionyl proteins account for about
65% of all new protein crystal structures phased by MAD. Selenium in
place of sulfur leads to only minimal changes in geometry and
hydrophobicity and crystals of Se-labeled proteins exhibit a high
degree of isomorphism with their wild type counterparts. By comparison,
the impact of MAD for solving new protein structures is currently not
matched by a similar success in the determination of nucleic acid
crystal structures. Bromine can be selectively introduced into
oligonucleotides in the form of 5-bromo-uracyl. However, applications
of MAD on bromo-derivatives are often not successful in practice,
probably because of base-stacking disruption and other structural
perturbations caused by bromo-derivatization. Crystallizability is
another issue with bromine derivatives: Not all halogen derivatives can
be crystallized under native conditions, and the derivative crystals do
not always diffract as well as the native ones. In addition, a possible
problem associated with halogen derivatives is that these halogenated
nucleotides are light sensitive; long-time exposure to X-ray or UV
sources may cause decomposition.
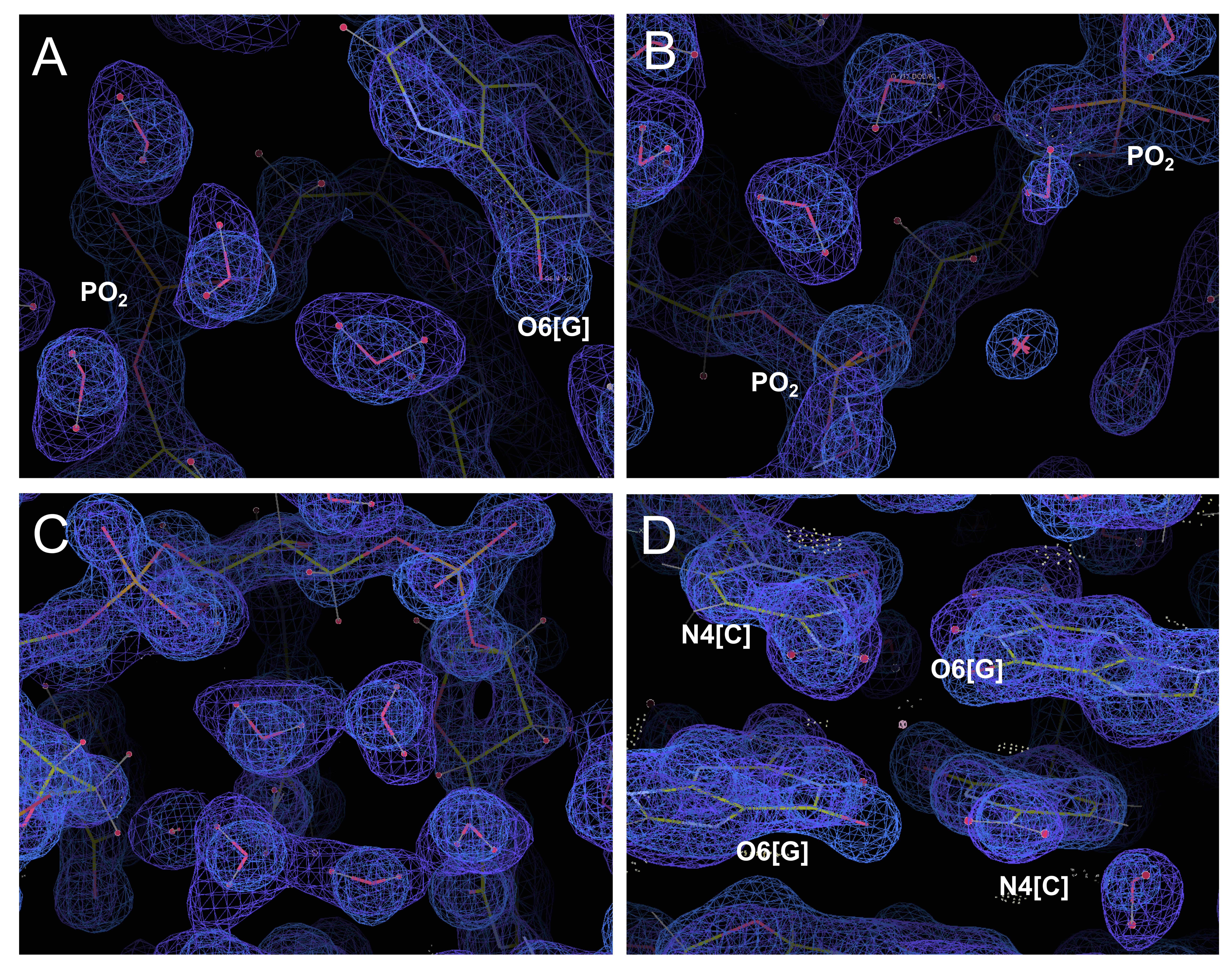
We have recently initiated a
program to investigate chemical synthetic routes for covalently
incorporating selenium into DNA. Several oxygen centers can potentially
be replaced by selenium (i.e. the 2-oxygen in pyrimidines, the ribose
2'-, 3'-, 4'- and 5'-oxygens and the non-bridging phosphate oxygen). We
have demonstrated that incorporation of 2'-selenomethyl-U into DNA
allows structure determination via MAD (c.v. refs. 77, 80). This
approach is also suitable for RNA structure determination. Moreover, we
have obtained initial experimental evidence that replacement of one of
the non-bridging phosphate oxygens by selenium and separation of the
resulting diastereoisomeric phosphoroseleneoates may furnish a
universal method for phasing X-ray diffraction data of native and
chemically modified nucleic acids as well as protein-nucleic acid
complexes (by labeling the nucleic acid instead of the protein) (c.v.
ref. 83; see the figure above, depicting a MAD-based electron density
map based on the PSe phasing approach).
|
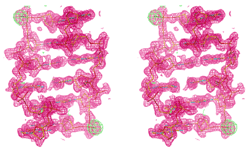 |