


 LIGPLOT Operating Manual
LIGPLOT Operating Manual
LIGPLOT Operating Manual
The plots produce by LIGPLOT can be improved in one of three ways.
a. Using LigEd
The preferred option for editing LIGPLOT plots is to use LigEd, a
Java-based LIGPLOT editor. You should have been sent decryption and
installation instructions for LigEd with your LIGPLOT
decryption code.
LigEd is supplied with Operating Instructions.
b. Editing the PostScript file
If you are familiar with PostScript files, you can make
simple amendments to the plot produced by LIGPLOT by editing the
ligplot.ps file.
The file is an ASCII text file, and so can be modified using any text editor. The sorts of amendments you can make are: changes to labels (in terms of size, colour and text), addition of other text, changes to colours, sizes, etc.
Some changes, of course, can be made simply by altering the
ligplot.prm parameter file (see section 5)
and re-running LIGPLOT.
c. Editing the ligplot.pdb file using interactive computer graphics
software
For more radical changes (say to change the positions or orientations of
sidechains/residues on the plot), you can use standard interactive computer
graphics software, such as QUANTA or Sybyl, to edit the output
ligplot.pdb file.
The ligplot.pdb file contains the coordinates of the flattened molecules, exactly as seen on the plot. You can read the file in as a standard PDB file and then use standard molecular modelling operations to modify the structure in any way that will make the final plot clearer.
Of course, the structure will be completely flat and the software might join some non-bonded atoms together by bonds simply because of their proximity to one another. But you can always break the bonds if this will make things clearer.
For example, if you are using QUANTA, you might modify the plot as follows:-
Note that the residues corresponding to hydrophobic contacts will be represented by one (or sometimes more) single carbon atoms. These, too, can be moved around the screen to more favourable positions.
Note that, when the ligplot.pdb file is saved by QUANTA, any blank chain ID's are replaced by the chain identifier "A". In this case, the residues in the ligplot.pdb file will no longer match the data in the ligplot.hhb, ligplot.nnb and ligplot.bonds files.Thus you will need to edit ligplot.pdb to convert the chain "A" back to chain " " (blank).
Note also, that when LIGPLOT plots the new diagram it uses the
information in the ligplot.bonds file to decide where, and of what
type, all the bonds are. So it does not matter how many bonds you broke or
created in QUANTA, as this information is not stored anywhere.
d. Altering the minimization parameters
The final method of improving a plot is to rerun LIGPLOT, or rather
LIGONLY, with different minimization parameters. These can be found
at the end of the parameter file, ligplot.prm.
In particular, if the minimization doesn't seem to have been able to run its course, you can change the parameter defining the number of loops for the minimization process.
By upweighting or downweighting certain parameters, you might be able to get closer to the plot you are after - but it is likely to be a very hit-and-miss business, and not one that is recommended.
e. Same protein, different ligands
Sometimes you may want to generate two or more LIGPLOTs for the same
protein but with different ligands bound. Direct comparison of such plots
can be hampered by the fact that LIGPLOT will probably have arranged
the residues in completely different positions on each plot in its efforts
to minimise the atom clashes and bond overlaps.
In such cases, it is more useful if the residues on the two (or more) plots are in equivalent positions relative to the ligand. This can be achieved using the ligplot.rcm file which is created whenever you run the program.
The ligplot.rcm file lists the centres of mass (x-y coords) of the residues on the plot (see Appendix B - LIGPLOT file formats). These can be used to restrain the centre-of-mass positions of the residues on a related plot, as follows:
The ligplot.rcm file generated for the above example is:
Res. Res Flattened
Atom Name Num coordinates
---- --- ----- ----------------
RESDUE CofM GLY C 250 -0.696 3.692
RESDUE CofM ALA C 251 0.805 0.245
RESDUE CofM TRP C 252 -3.097 -1.856
RESDUE CofM HIS - 57 6.414 -3.497
RESDUE CofM GLY - 216 -4.914 3.844
RESDUE CofM ASP - 102 6.205 -8.482
RESDUE CofM SER - 195 -1.501 -8.050
RESDUE CofM SER - 214 5.391 0.946
RESDUE CofM TRP - 215 -9.097 1.708
RESDUE CofM SER - 217 -9.342 -2.440
RESDUE CofM GLY - 193 -5.669 -6.010
RESDUE CofM SER - 190 -13.399 0.649
RESDUE CofM CYS - 191 -8.321 -9.347
RESDUE CofM GLY - 226 -7.737 6.593
RESDUE CofM MET - 192 -11.215 -5.617
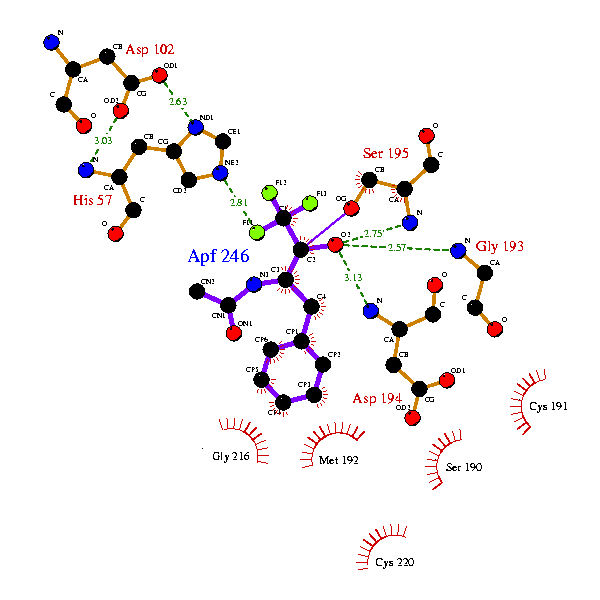
The above example might now become:
Res. Res Flattened
Atom Name Num coordinates
---- --- ----- ----------------
RESDUE CofM APF - 246 0.000 0.000
RESDUE CofM HIS - 57 6.414 -3.497
RESDUE CofM ASP - 102 6.205 -8.482
RESDUE CofM SER - 195 -1.501 -8.050
RESDUE CofM GLY - 193 -5.669 -6.010
RESDUE CofM CYS - 191 -8.321 -9.347
RESDUE CofM SER - 190 -13.399 0.649
RESDUE CofM MET - 192 -11.215 -5.617
Although this gives the residuesin roughly equivalent position, you can see in this example, that the ligand isn't quite in the equivalent orientation, and some of the side-chains of the protein, too, are in different orientations.
To improve on the plot, you can add atomic restraints. These take the form of ATOM records - which have exactly the same format as ATOM records in a PDB file (see Appendix A - Brookhaven file format).
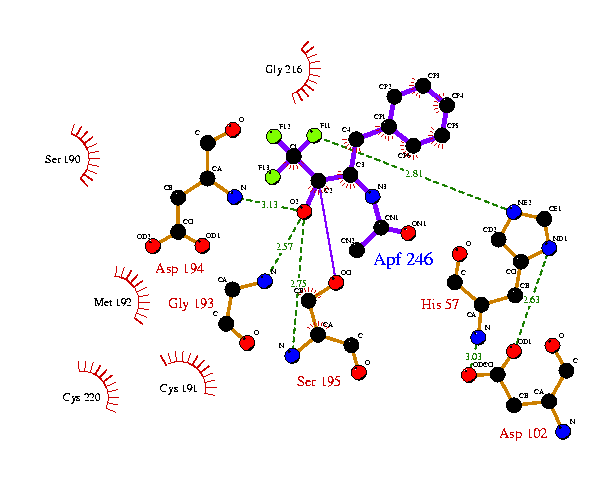
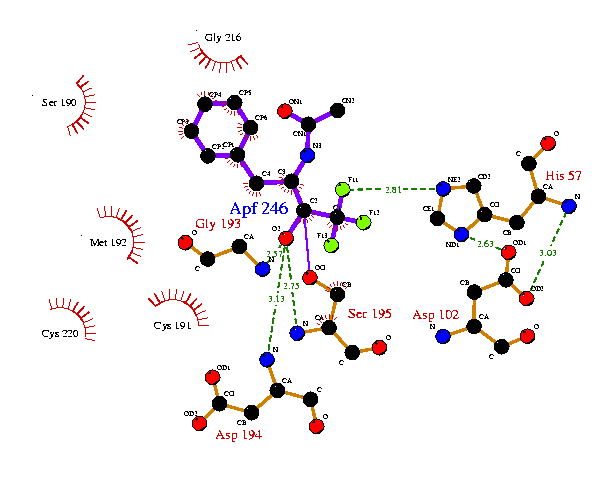
Consider, for example, the His57 and Asp102 residues in the plots above. Although these residues are in equivalent positions it would be nice to have them oriented in the same manner. This can be done by removing the ATOM records from the ligplot.pdb file generated by the 8gch plot and inserting them in the 6gch.rcm file as follows:-
Res. Res Flattened
Atom Name Num coordinates
---- --- ----- ----------------
RESDUE CofM APF - 246 0.000 0.000
ATOM 385 N HIS 57 7.920 -4.629 0.000 1.00 6.92 6.920
ATOM 386 CA HIS 57 8.002 -3.175 0.000 1.00 7.11 7.110
ATOM 387 C HIS 57 9.316 -2.413 0.000 1.00 7.57 7.570
ATOM 388 O HIS 57 9.345 -1.154 0.000 1.00 8.14 8.140
ATOM 389 CB HIS 57 6.615 -2.482 0.000 1.00 7.18 7.180
ATOM 390 CG HIS 57 5.474 -3.458 0.000 1.00 7.22 7.220
ATOM 391 ND1 HIS 57 5.557 -4.819 0.000 1.00 6.95 6.950
ATOM 392 CD2 HIS 57 4.134 -3.163 0.000 1.00 6.92 6.920
ATOM 393 CE1 HIS 57 4.330 -5.324 0.000 1.00 7.49 7.490
ATOM 394 NE2 HIS 57 3.448 -4.350 0.000 1.00 7.23 7.230
ATOM 729 N ASP 102 3.849 -9.704 0.000 1.00 7.78 7.780
ATOM 730 CA ASP 102 5.273 -9.283 0.000 1.00 6.95 6.950
ATOM 731 C ASP 102 6.476 -10.179 0.000 1.00 7.25 7.250
ATOM 732 O ASP 102 7.641 -9.702 0.000 1.00 7.16 7.160
ATOM 733 CB ASP 102 5.268 -7.731 0.000 1.00 7.18 7.180
ATOM 734 CG ASP 102 6.683 -7.212 0.000 1.00 6.68 6.680
ATOM 735 OD1 ASP 102 7.611 -8.064 0.000 1.00 7.10 7.100
ATOM 736 OD2 ASP 102 6.838 -5.976 0.000 1.00 7.75 7.750
RESDUE CofM SER - 195 -1.501 -8.050
RESDUE CofM GLY - 193 -5.669 -6.010
RESDUE CofM CYS - 191 -8.321 -9.347
RESDUE CofM SER - 190 -13.399 0.649
RESDUE CofM MET - 192 -11.215 -5.617
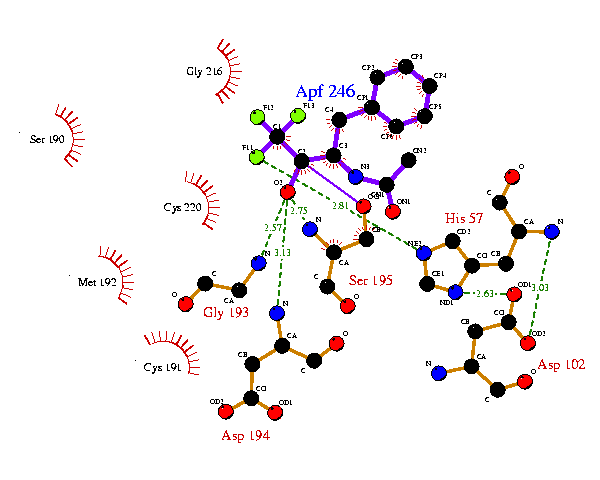
This now gets the His57 and Asp102 residues as on the 8gch plot, but the ligand orientation is still not right. So equivalent atom-positions from the 8gch ligplot.pdb file can be taken to fix the ligand. Similarly, the other sidechains can be further tied down with reference to the 8gch plot. The final result might be:
This now bears a much closer resemblance to the equivalent plot for 8gch. The 6gch.rcm file that gave this plot was:-
Res. Res Flattened
Atom Name Num coordinates
---- --- ----- ----------------
ATOM 1782 F11 APF 246 0.023 -4.549 0.000 0.30 14.18 14.180
ATOM 1784 F13 APF 246 -2.105 -3.859 0.000 1.00 13.79 13.790
ATOM 1788 CP4 APF 246 -6.701 -0.070 0.000 1.00 15.35 15.350
ATOM 385 N HIS 57 7.920 -4.629 0.000 1.00 6.92 6.920
ATOM 386 CA HIS 57 8.002 -3.175 0.000 1.00 7.11 7.110
ATOM 387 C HIS 57 9.316 -2.413 0.000 1.00 7.57 7.570
ATOM 388 O HIS 57 9.345 -1.154 0.000 1.00 8.14 8.140
ATOM 389 CB HIS 57 6.615 -2.482 0.000 1.00 7.18 7.180
ATOM 390 CG HIS 57 5.474 -3.458 0.000 1.00 7.22 7.220
ATOM 391 ND1 HIS 57 5.557 -4.819 0.000 1.00 6.95 6.950
ATOM 392 CD2 HIS 57 4.134 -3.163 0.000 1.00 6.92 6.920
ATOM 393 CE1 HIS 57 4.330 -5.324 0.000 1.00 7.49 7.490
ATOM 394 NE2 HIS 57 3.448 -4.350 0.000 1.00 7.23 7.230
ATOM 729 N ASP 102 3.849 -9.704 0.000 1.00 7.78 7.780
ATOM 730 CA ASP 102 5.273 -9.283 0.000 1.00 6.95 6.950
ATOM 731 C ASP 102 6.476 -10.179 0.000 1.00 7.25 7.250
ATOM 732 O ASP 102 7.641 -9.702 0.000 1.00 7.16 7.160
ATOM 733 CB ASP 102 5.268 -7.731 0.000 1.00 7.18 7.180
ATOM 734 CG ASP 102 6.683 -7.212 0.000 1.00 6.68 6.680
ATOM 735 OD1 ASP 102 7.611 -8.064 0.000 1.00 7.10 7.100
ATOM 736 OD2 ASP 102 6.838 -5.976 0.000 1.00 7.75 7.750
ATOM 1370 N GLY 193 -3.838 -6.317 0.000 1.00 7.53 7.530
ATOM 1371 CA GLY 193 -5.074 -5.514 0.000 1.00 6.95 6.950
ATOM 1372 C GLY 193 -6.327 -6.383 0.000 1.00 6.51 6.510
ATOM 1373 O GLY 193 -7.437 -5.828 0.000 1.00 6.00 6.000
RESDUE CofM CYS - 191 -8.321 -9.347
RESDUE CofM SER - 190 -13.399 0.649
RESDUE CofM MET - 192 -11.215 -5.617
Further refinement is still possible, including modifying the coordinates of individual sidechains, say by manipulating them using a graphics program as described in section b above.
LIGPLOT Operating Manual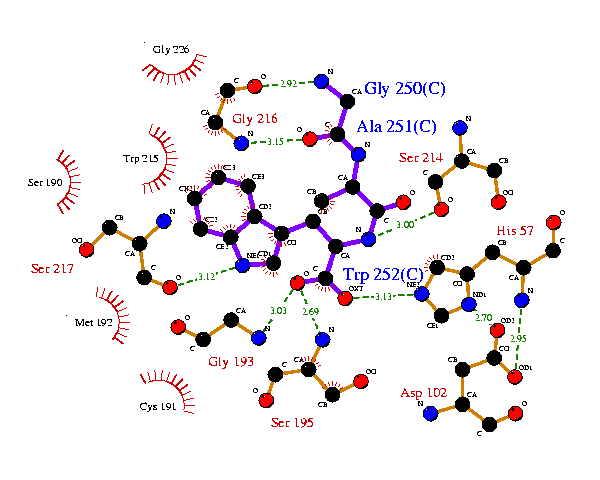{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}