 | |||||||||||||||
 |
 |
 |
|
 |
|
 |
|
 |
|
 |
|
 |
|
 |
 |
AMBER Archive (2009)
Subject: [AMBER] aromatic ring getting distorted
From: Jio M (jiomm_at_yahoo.com)
Date: Fri Dec 04 2009 - 04:03:49 CST
- Previous message: intra\\sa175950: "RE: [AMBER] Comparison of the CHARMM and AMBER parameters for alkane"
- Next in thread: case: "Re: [AMBER] aromatic ring getting distorted"
- Reply: case: "Re: [AMBER] aromatic ring getting distorted"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
Dear Amber users;
I have a ring system like shown in two attached files which is to be
used as ligands.When I run dynamics the ring system get distorted more
or less like cyclohexane ring. This should NOT happen according to atom
types.
I tried by changing atom type cc (cc.png attached) to cp (cp.png
attached) so that they correspond to carbon atoms attaching two rings.
But when I run dynamics (imin=0) Amber is considering rings to be
cyclohexane like not aromatic though I have not changed the atom types
of other atoms.
Should I define bond-order by myself and also should I change atom type from cc to cp?
thanks and regards;
JIomm
- image/png attachment: cc.png
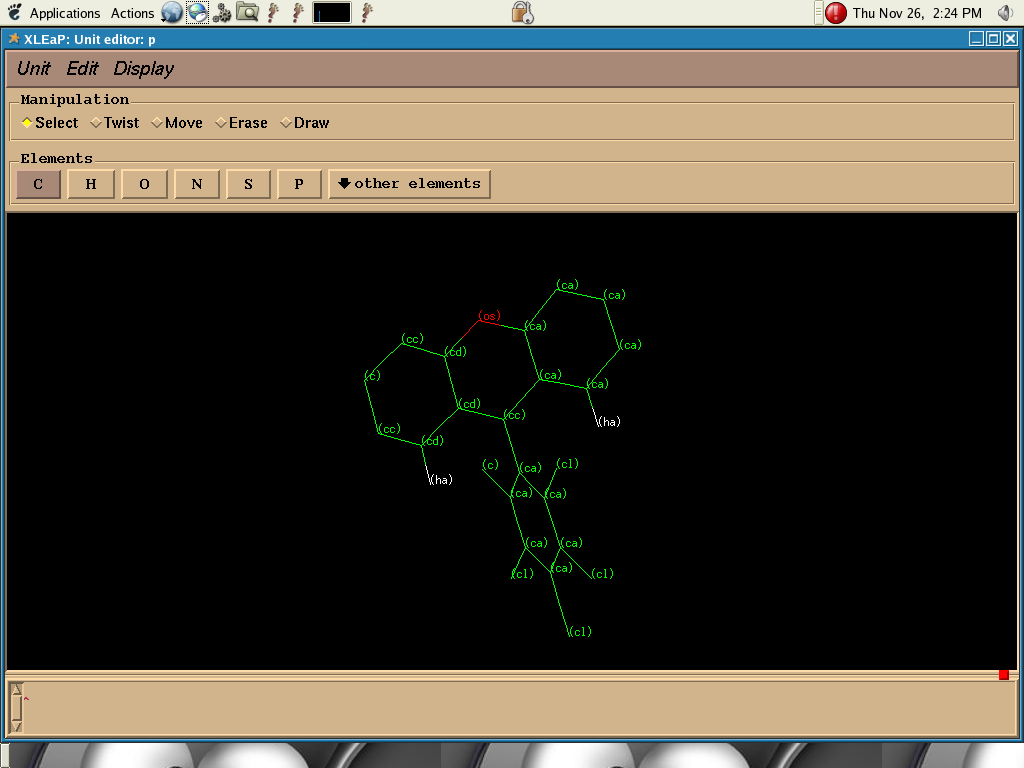{kind=link}
- image/png attachment: cp.png
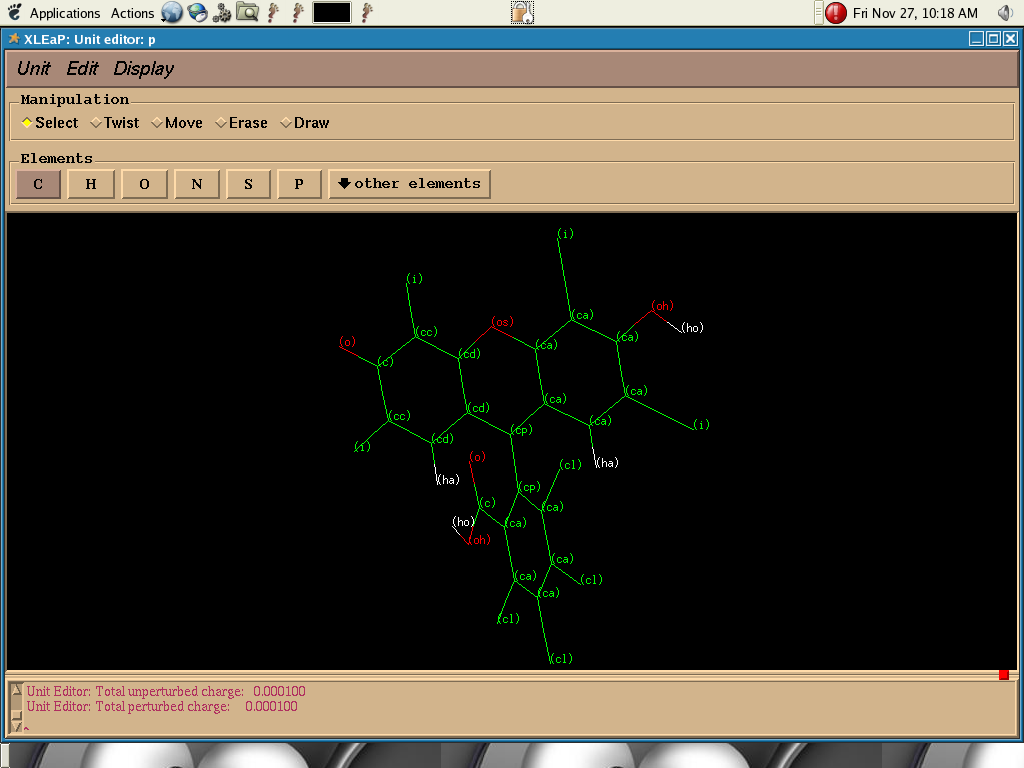{kind=link}
_______________________________________________
AMBER mailing list
AMBER_at_ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
- Previous message: intra\\sa175950: "RE: [AMBER] Comparison of the CHARMM and AMBER parameters for alkane"
- Next in thread: case: "Re: [AMBER] aromatic ring getting distorted"
- Reply: case: "Re: [AMBER] aromatic ring getting distorted"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
|
VU Home |
VUMC Home |
People Finder |
University Calendar
webmaster- modified on December 06, 2009 |