 | |||||||||||||||
 |
 |
 |
|
 |
|
 |
|
 |
|
 |
|
 |
|
 |
 |
AMBER Archive (2009)
Subject: Re: [AMBER] how to know that adequate equilibration from rmsd curve of equilibration
From: Naser Alijabbari (na3m_at_virginia.edu)
Date: Sat Jun 27 2009 - 13:19:23 CDT
- Previous message: Cihan Aydin: "Re: [AMBER] how to know that adequate equilibration from rmsd curve of equilibration"
- In reply to: Cihan Aydin: "Re: [AMBER] how to know that adequate equilibration from rmsd curve of equilibration"
- Next in thread: Cihan Aydin: "Re: [AMBER] how to know that adequate equilibration from rmsd curve of equilibration"
- Reply: Cihan Aydin: "Re: [AMBER] how to know that adequate equilibration from rmsd curve of equilibration"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
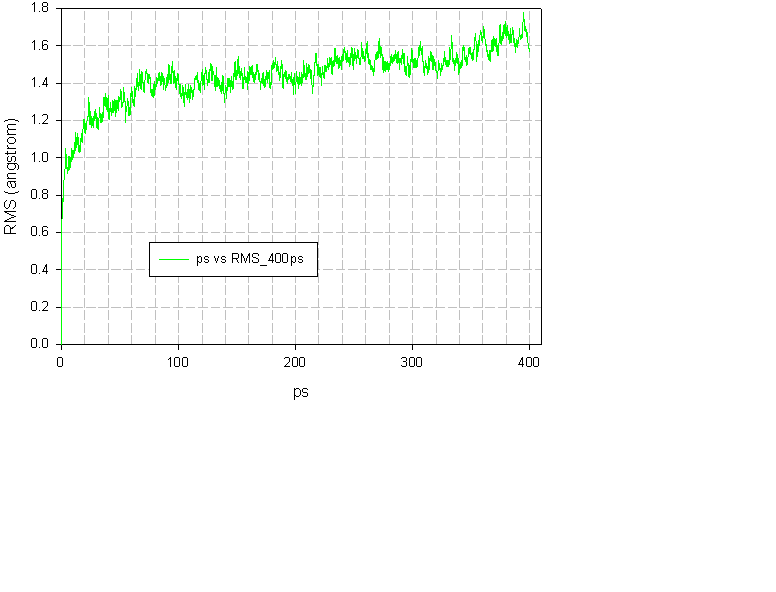
I have attached the RMSd plot from a 400ps NVE simulation of a protein with
108 residue. The plot is similar to the one in the paper but when I plot the
eigenfrequencies (from quasiharmonic analysis) every 10ps, around 350ps
something, which I don't know how to characterize, happens and the
eigenfrequency which were similar between previous 10ps intervals, change
dramatically. So ETOT and RMSd might not be enough to know that something
odd is happening.
On Sat, Jun 27, 2009 at 10:29 AM, Cihan Aydin <cihan.aydin_at_umassmed.edu>wrote:
> I encountered this e-mail while roaming on the reflector. I'll cite a
> paper that helped me much.
>
> Stella,L.; Melchionna,S.
> J. of Chem. Phys., Vol. 109, No.23, 1998
>
> Cheers,
>
> Cihan
>
>
> On Thu, 2009-05-28 at 14:15 -0400, Carlos Simmerling wrote:
> > that's way too general. it depends on many things, such as the quality of
> > the experimental structure, the difference in conditions between your
> > simulation and the experiment (pH, solvent, ions etc) and where in the
> > structure the rmsd values come from. rmsd is a very broad measure and a
> > value of 2 may mean that all regions differ by about 2, or that many
> differ
> > by 1 and others differ by a larger number (for example 5, or maybe 10).
> rmsd
> > itself is only 1 measure of simulation quality so I strongly disagree
> that
> > simulation rmsd values should "not be greater than 2". if you do get
> larger
> > values, though, you should find out why and see if there is a way to
> > validate that the behavior is reasonable.
> >
> > equilibration can be very tricky and there is no one "correct" protocol
> we
> > can give. it depends again on the initial structure and any differences
> in
> > your simulation conditions from the expt. it is entirely possible to have
> > most of the structure look ok, but due to bad placement of an H atom or
> even
> > an ion, part of the initial structure will be incorrectly modeled and the
> > simulation will not give a good model. equilibration can help with these
> but
> > you need detailed information about differences between MD and experiment
> to
> > proceed. we tend to be cautious and use about 10 steps in our general
> > equilibration procedure, and more when anything was modeled (such as a
> side
> > chain or a ligand).
> >
> >
> > On Thu, May 28, 2009 at 2:08 PM, nicholus bhattacharjee <
> > nicholusbhattacharjee_at_gmail.com> wrote:
> >
> > > Thank you Bill. But you did not answer my second question. Is the rmsd
> > > value
> > > between the initial structure and the final structure be not more than
> 2
> > > Armstrong.
> > > _______________________________________________
> > > AMBER mailing list
> > > AMBER_at_ambermd.org
> > > http://lists.ambermd.org/mailman/listinfo/amber
> > >
> > _______________________________________________
> > AMBER mailing list
> > AMBER_at_ambermd.org
> > http://lists.ambermd.org/mailman/listinfo/amber
> >
> --
> Cihan Aydin
> UMass Graduate School of Biomedical Sciences
> PhD Student @ Schiffer Lab
>
> 364 Plantation St. LRB 970M
> Worcester, MA 01605
>
> cihan.aydin_at_umassmed.edu
> +1 (508) 856-3430
>
>
> _______________________________________________
> AMBER mailing list
> AMBER_at_ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
- image/bmp attachment: example_run.bmp
{kind=link}
_______________________________________________
AMBER mailing list
AMBER_at_ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
- Previous message: Cihan Aydin: "Re: [AMBER] how to know that adequate equilibration from rmsd curve of equilibration"
- In reply to: Cihan Aydin: "Re: [AMBER] how to know that adequate equilibration from rmsd curve of equilibration"
- Next in thread: Cihan Aydin: "Re: [AMBER] how to know that adequate equilibration from rmsd curve of equilibration"
- Reply: Cihan Aydin: "Re: [AMBER] how to know that adequate equilibration from rmsd curve of equilibration"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
|
VU Home |
VUMC Home |
People Finder |
University Calendar
webmaster- modified on June 28, 2009 |