 | |||||||||||||||
 |
 |
 |
|
 |
|
 |
|
 |
|
 |
|
 |
|
 |
 |
AMBER Archive (2009)
Subject: [AMBER] REMD fixed atoms
From: Maxime Louet (maxime.louet_at_etudiant.univ-rennes1.fr)
Date: Fri Jan 30 2009 - 11:26:08 CST
- Previous message: David A. Case: "Re: [AMBER] Dr. Ramesh here"
- Next in thread: Carlos Simmerling: "Re: [AMBER] REMD fixed atoms"
- Reply: Carlos Simmerling: "Re: [AMBER] REMD fixed atoms"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
Dear all,
I am a new user of Amber 9 and I'm trying to simulate a small peptide
dimers using REMD, FF96, igb=5 and no restraint.
However, I have some problems. The calculations works fine (without
any error message), but when I analyse the trajectory of any single
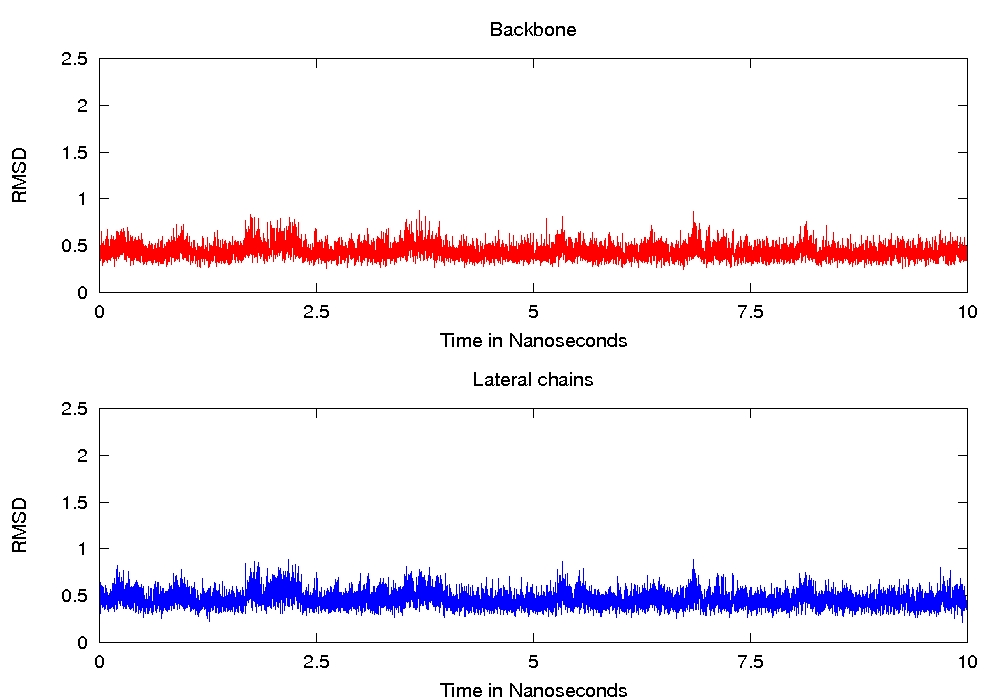
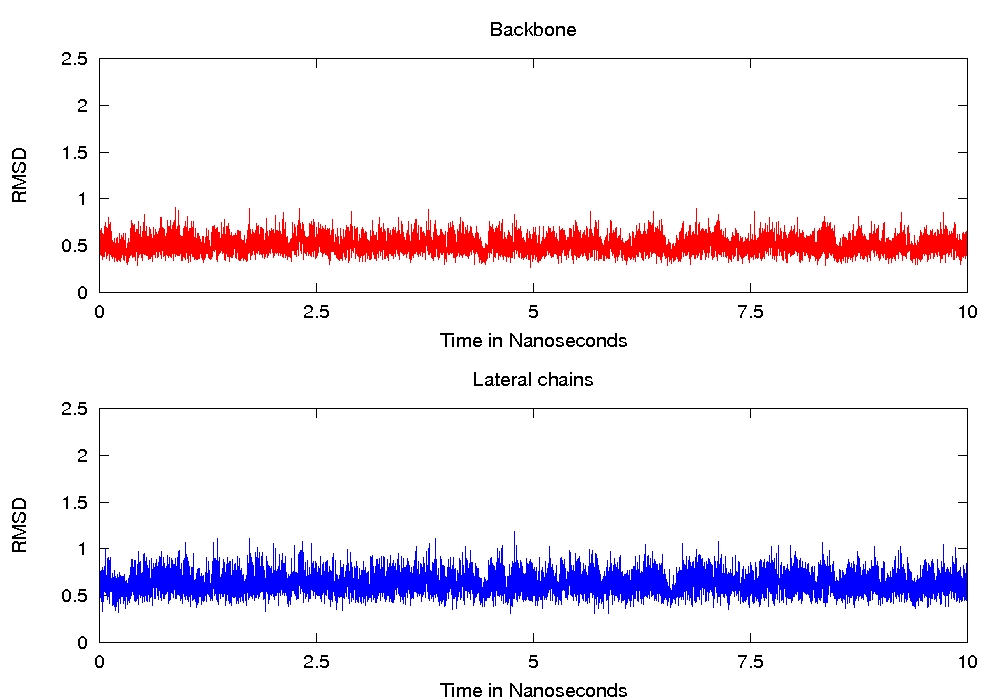
replica, it seems that the backbones and lateral chains doesn't move
(even at high temperature), see attached files for the RMSD of replica
1 and 16.
Using the same kind of input with regular sander (no REMD) lead to
totally "normal" simulation. Does anybody have an answer ?
I used 16 replicas with 32 processors, temperatures are between 190K and 503K.
Here is one of my input for the replica starting with T=190 :
&cntrl
imin=0, irest=1, ntx=5,
nstlim=1000, dt=0.002,
ntc=2, ntf=2,
ntt=3, gamma_ln=50,
tempi=0.0, temp0=190.0,
ntpr=500, ntwx=500,
ntb=0, igb=5,
gbsa=1, saltcon=0.15,
cut=25.0, rgbmax=25.0,
repcrd=1, numexchg=12500,
/
Thank you in advance, for the time you will spend to answer my question.
Maxime Louet
_______________________________________________
AMBER mailing list
AMBER_at_ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
- Previous message: David A. Case: "Re: [AMBER] Dr. Ramesh here"
- Next in thread: Carlos Simmerling: "Re: [AMBER] REMD fixed atoms"
- Reply: Carlos Simmerling: "Re: [AMBER] REMD fixed atoms"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
|
VU Home |
VUMC Home |
People Finder |
University Calendar
webmaster- modified on February 01, 2009 |