 | |||||||||||||||
 |
 |
 |
|
 |
|
 |
|
 |
|
 |
|
 |
|
 |
 |
AMBER Archive (2008)
Subject: RE: AMBER: reg unstability of structure in md
From: balaji nagarajan (balaji_sethu_at_hotmail.com)
Date: Thu Dec 04 2008 - 22:32:21 CST
- Next message: N.R. Jena: "Re: AMBER: reg unstability of structure in md"
- Previous message: N.R. Jena: "Re: AMBER: MM/GBSA Error during deccomposition"
- In reply to: Carlos Simmerling: "Re: AMBER: reg unstability of structure in md"
- Next in thread: N.R. Jena: "Re: AMBER: reg unstability of structure in md"
- Reply: N.R. Jena: "Re: AMBER: reg unstability of structure in md"
- Reply: Carlos Simmerling: "Re: AMBER: reg unstability of structure in md"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
dear amber ,
I am doing md for a junction structure .,
its solved by xray diffraction and i am using that pdb for dynamics
no ions were found in the structure and the pH was 7 during crystallization .,
and no ligands present in it .,
actually on doing minimization with truncated octahedralbox of water
the water box and the dna are perfect .
I did equlibration and md as in the tutorial for poly - AT
but when i analysed the results as mentioned there .,
i am getting the perfect plots
but when i view the trajectories in vmd
its giving a picture like ( i have attached ) this .
what could be the reason for it
thanks in advance
balaji.,
Date: Thu, 4 Dec 2008 06:28:12 -0500
From: carlos.simmerling_at_gmail.com
To: amber_at_scripps.edu
Subject: Re: AMBER: reg unstability of structure in md
we'll need more information, such as where the pdb file came from (experiment? what type, and how well defined is the structure? were any regions missing?), if there are experimental measures of stability, if ions are important, if pH is important, if there are any non-standard force fields involved (such as a ligand), and then we need to know more about the equilibration procedure that you used. finally, explain what you mean by "it went bad". what analysis did you do?
On Wed, Dec 3, 2008 at 11:17 PM, balaji nagarajan <balaji_sethu_at_hotmail.com> wrote:
dear amber ,
I am a new user to amber ,
I changed my pdb format and loaded in xleap its loaded ..,
and i am sure my pdb is quite in a right form isn't it .,
it gave a message in terminal like this ...
dna1 = loadpdb "j0.pdb"
Loading PDB file: ./j0.pdb
total atoms in file: 808
Leap added 448 missing atoms according to residue templates:
448 H / lone pairs
I solvated the structure in water (TIP3P)
by giving
solvateoct model2 TIP3PBOX 8.0
and i viewed the pdb its good
but when i do the minimization and the procedure as given in poly-AT tutorial
in explicit water for 120 ps ..
my molecule is no more stable it went bad
what could be the reason
help me out to solve the problem
thanks in advance .....
Some big movies will hit the screen this Christmas. Catch all the previews and promos on MSN Entertainment Try it!
_________________________________________________________________
Register once and play all contests. Increase your scores with bonus credits for logging in daily on MSN.
http://specials.msn.co.in/msncontest/index.aspx
- image/png attachment: md_1crdwithwaterbox.png
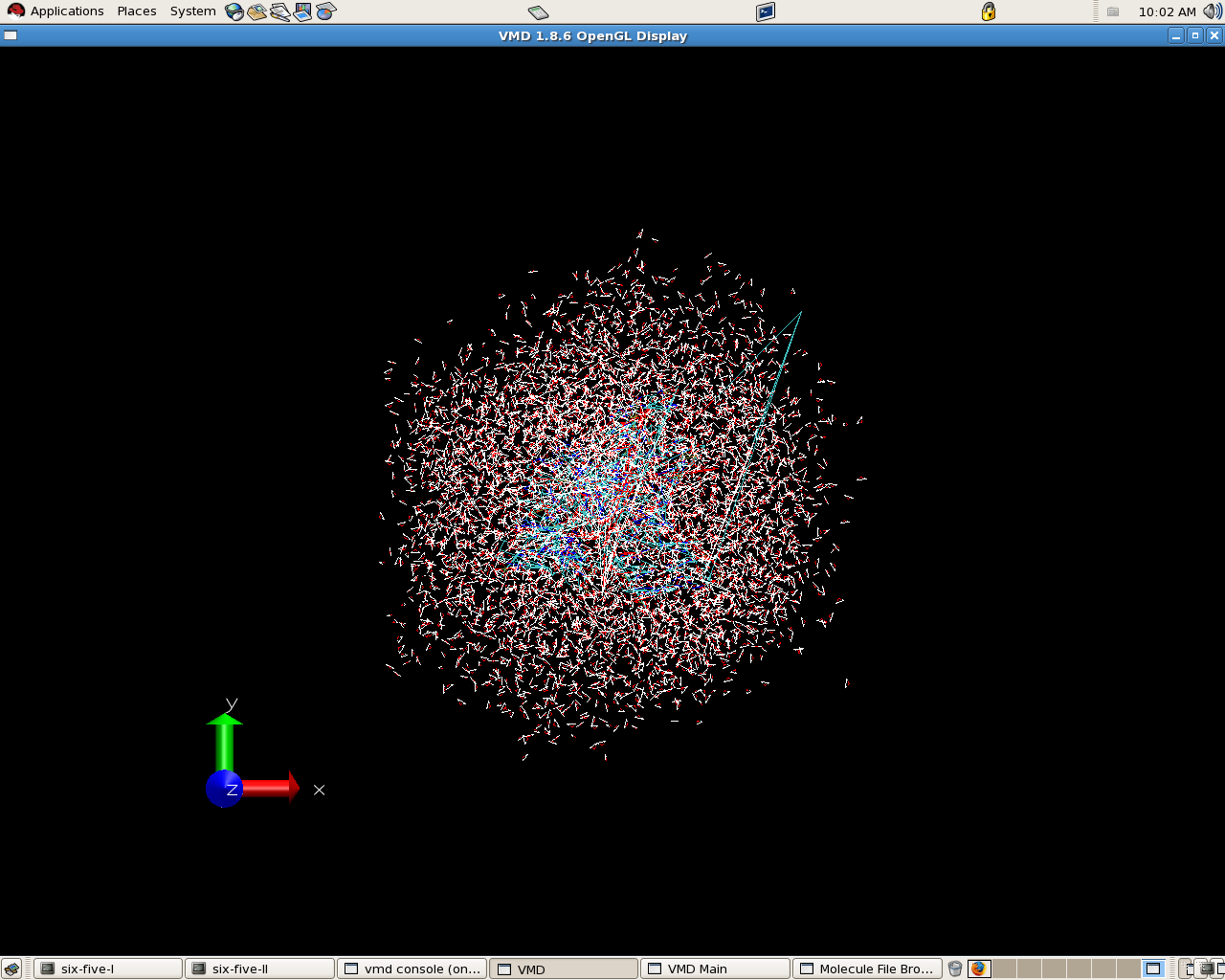{kind=link}
- image/png attachment: molecule.png
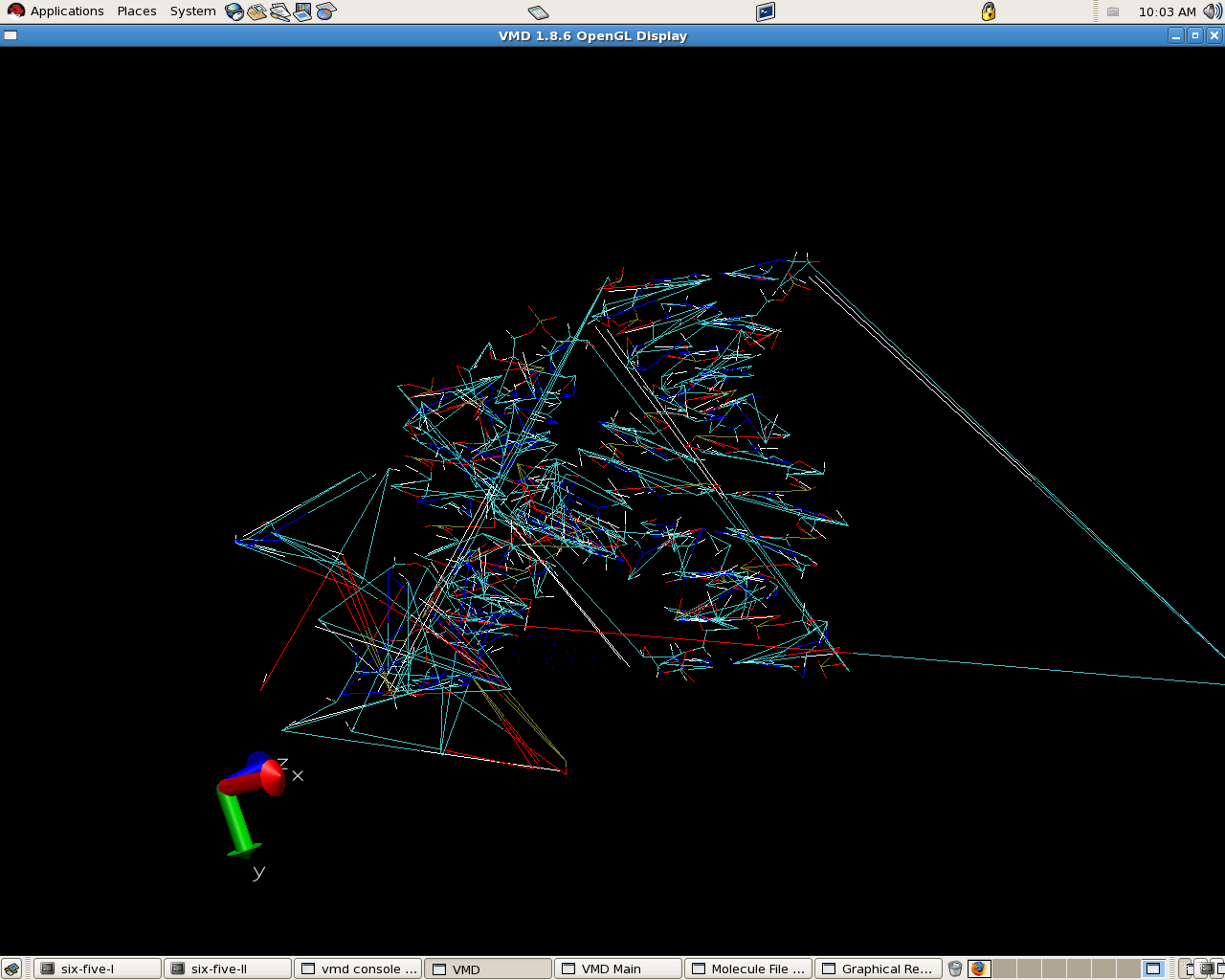{kind=link}
The AMBER Mail Reflector
To post, send mail to amber_at_scripps.edu
To unsubscribe, send "unsubscribe amber" (in the *body* of the email)
to majordomo_at_scripps.edu
- Next message: N.R. Jena: "Re: AMBER: reg unstability of structure in md"
- Previous message: N.R. Jena: "Re: AMBER: MM/GBSA Error during deccomposition"
- In reply to: Carlos Simmerling: "Re: AMBER: reg unstability of structure in md"
- Next in thread: N.R. Jena: "Re: AMBER: reg unstability of structure in md"
- Reply: N.R. Jena: "Re: AMBER: reg unstability of structure in md"
- Reply: Carlos Simmerling: "Re: AMBER: reg unstability of structure in md"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
|
VU Home |
VUMC Home |
People Finder |
University Calendar
webmaster- modified on January 30, 2009 |