 | |||||||||||||||
 |
 |
 |
|
 |
|
 |
|
 |
|
 |
|
 |
|
 |
 |
AMBER Archive (2008)
Subject: Re: AMBER: Non bond list error
From: Robert Duke (rduke_at_email.unc.edu)
Date: Thu Oct 23 2008 - 15:44:29 CDT
- Next message: Wang,Ying: "Re: AMBER: Non bond list error"
- Previous message: Gustavo Seabra: "Re: AMBER: pdb structure is only half the molecule."
- In reply to: Robert Duke: "Re: AMBER: Non bond list error"
- Next in thread: neville forlemu: "Re: AMBER: Non bond list error"
- Reply: neville forlemu: "Re: AMBER: Non bond list error"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
Actually, I noticed I said "you overflowed the counter", and then show that
you didn't... (oh, oops). So it is memory corruption. What I don't
understand is why you are not dying with some sort of "out of memory" error
from sander, associated with asking for more memory than is available. For
pmemd, anywhere I allocate dynamic memory, I check for a success return
code, so the way you should experience running out of memory there is to get
an explicit error message. Because sander has a preallocated memory pool
strategy, I suspect that other things are possible... Bottom line on all
this - I think it is a good idea to not run more than roughly 100,000 atoms
on a single processor, especially for sander. And if you run it on 4
processors but they all share the same limited physical memory, you may also
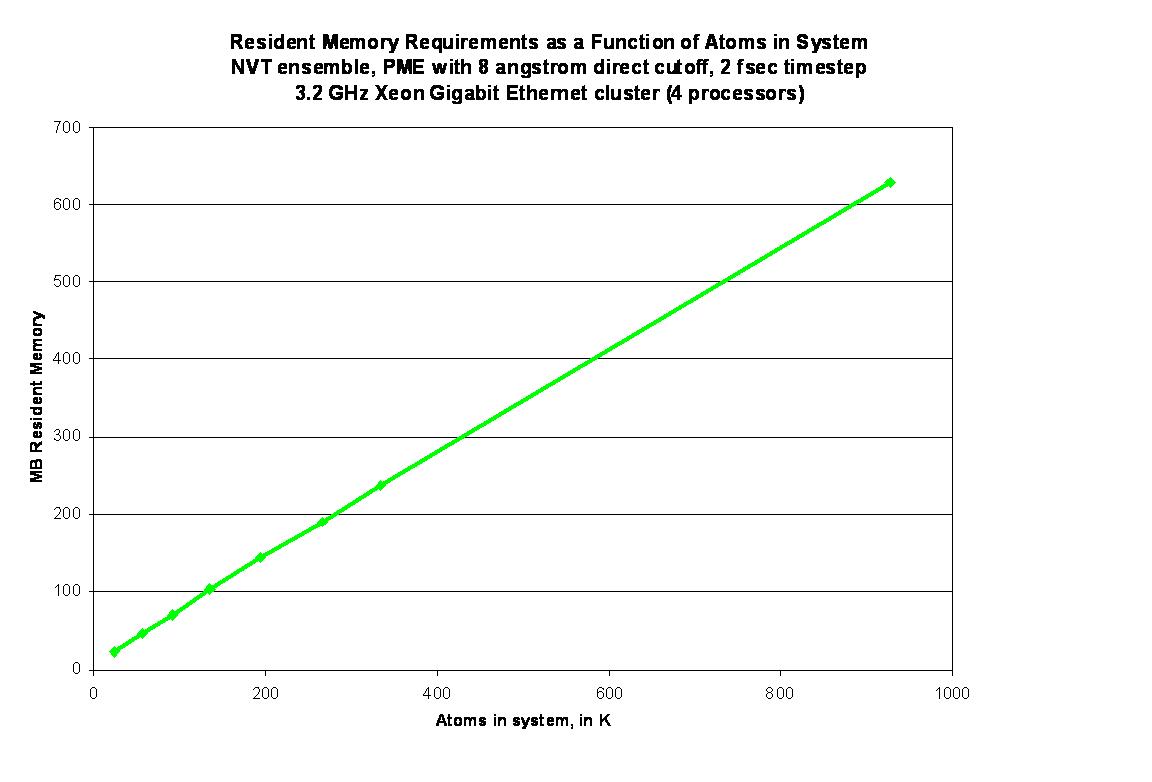
hit trouble. I attached a graphic on pmemd memory requirements - a jpg so
it should be widely viewable. My rule of thumb for pmemd is that 4
processors, each with 1 GB of actual physical memory, can handle up to 1
million atoms with the default 8 angstrom cutoff. Sander will take more.
There are also buffer space considerations in an mpi application (within mpi
itself, not in the app), that further muddy the waters, but following this
guideline you should be safe.
Regards - Bob
----- Original Message -----
From: "Robert Duke" <rduke_at_email.unc.edu>
To: <amber_at_scripps.edu>
Sent: Thursday, October 23, 2008 4:14 PM
Subject: Re: AMBER: Non bond list error
> As Ross will tell you too:
> 1) Don't increase cut to 12, leave it at the default (of 8)
> 2) Run this on at least 4 processors using the MPI version of pmemd or
> sander (I know you are using sander here; pmemd requires less memory).
> Even higher processor counts will reduce your risk of memory overflow
> further. Your pairlist went negative because you incremented it past a 31
> bit digit; with the commonly used integer format on computers these days
> (twos-complement), this results in a negative number (and is clearly an
> error condition). Is this memory usage reasonable for the size problem
> you have? Well, that cutoff plus skin will produce about 552 pairs per
> atom. If you had 1,000,000 atoms (and you are close), that would be
> 552,000,000 pairs. Not enough to overflow the list counter. BUT that is
> 552,000,000 pairs * 4 bytes per integer, means 2 GB in the nonbonded list
> alone. Most machines, you are pushing it to get much over 1.5 GB for the
> application (I have not looked recently, so that is off the top of my
> head). With true 32 bit executables, you are out of address space; with
> the newer 64 bit chips, you have bits to specify more than 2 GB of
> addresses, but you may not have enough actual memory. And remember that
> the pairlist is only part of your memory consumption. No resource is
> infinite on a computer...
> Regards - Bob Duke
> ----- Original Message -----
> From: "Wang,Ying" <wangying_at_ufl.edu>
> To: <amber_at_scripps.edu>
> Sent: Thursday, October 23, 2008 3:26 PM
> Subject: RE: AMBER: Non bond list error
>
>
>> Hi, Ross,
>>
>> Thanks a lot!
>>
>> My input file is as below:
>> 50ps MD with res
>> &cntrl
>> imin = 0,
>> irest = 0,
>> ntx = 1,
>> ntb = 1,
>> cut = 12,
>> ntr = 1,
>> ntc = 2,
>> ntf = 2,
>> tempi = 0.0,
>> temp0 = 300.0,
>> ntt = 3,
>> gamma_ln = 2.0,
>> nstlim = 50000, dt = 0.001
>> ntpr = 1000, ntwx = 1000, ntwr = 1000
>> nmropt=1
>> /
>> &wt TYPE='TEMP0', istep1=0, istep2=50000,
>> value1=0.1, value2=300.0, /
>> &wt TYPE='END' /
>> Keep system fixed with weak restraints
>> 20.0
>> RES 1 5076
>> END
>> END
>>
>> and the NPT is as below:
>>
>> NPT: 50ps MD
>> &cntrl
>> imin = 0, irest = 1, ntx = 7,
>> ntb = 2, pres0 = 1.0, ntp = 1,
>> taup = 2.0,
>> cut = 12, ntr = 1,
>> ntc = 2, ntf = 2,
>> tempi = 300.0, temp0 = 300.0,
>> ntt = 3, gamma_ln = 2.0,
>> nstlim = 50000, dt = 0.001,
>> ntpr = 1000, ntwx = 1000, ntwr = 1000
>> /
>> Keep fixed with weak restraints
>> 20.0
>> RES 217 954
>> END
>> Keep fixed with weak restraints
>> 20.0
>> RES 1909 2646
>> END
>> Keep fixed with weak restraints
>> 20.0
>> RES 3601 4338
>> END
>> res also
>> 5.0
>> RES 955 1692
>> END
>> res also
>> 5.0
>> RES 2647 3384
>> END
>> res also
>> 5.0
>> RES 4339 5076
>> END
>> END
>>
>>
>>
>> Thanks again!!!!!!!!!!!!!!!
>>
>>
>>
>> On Thu Oct 23 14:38:09 EDT 2008, Ross Walker <ross_at_rosswalker.co.uk>
>> wrote:
>>
>>> Hi Wang,
>>>
>>> 800K atoms is pretty large and while sander / pmemd should support this
>>> size
>>> (I think 999,999 is the limit right now due to file formatting) you may
>>> run
>>> into problems that haven't been seen before.
>>>
>>> It's not obvious what is going wrong in your case but the numbers don't
>>> make
>>> any sense (a negative capacity!) which suggests either memory corruption
>>> through an array overflow or the number of pairs is larger than a signed
>>> integer and is overflowing. Even at 800K atoms you shouldn't have this
>>> many
>>> pairs though. Can you post your input file so we can take a look? I
>>> suspect
>>> you have cut set too high or perhaps are not running PME etc.
>>>
>>> All the best
>>> Ross
>>>
>>>> -----Original Message-----
>>>> From: owner-amber_at_scripps.edu [mailto:owner-amber_at_scripps.edu] On
>>>> Behalf
>>>> Of Wang,Ying
>>>> Sent: Thursday, October 23, 2008 10:20 AM
>>>> To: amber_at_scripps.edu
>>>> Subject: AMBER: Non bond list error
>>>>
>>>> Hi, Dear AMBERs,
>>>>
>>>> I meet a problem when I run a simulation of a system consist of
>>>> 799889 atoms.
>>>>
>>>> * NB pairs 451 0 exceeds capacity (
>>>> -28510921) 7
>>>> SIZE OF NONBOND LIST = -28510921
>>>> SANDER BOMB in subroutine nonbond_list
>>>> Non bond list overflow!
>>>> check MAXPR in locmem.f
>>>>
>>>> Could anyone tell me what's happen?
>>>>
>>>> Thanks a lot!
>>>>
>>>> -----------------------------------------------------------------------
>>>> The AMBER Mail Reflector
>>>> To post, send mail to amber_at_scripps.edu
>>>> To unsubscribe, send "unsubscribe amber" (in the *body* of the email)
>>>> to majordomo_at_scripps.edu
>>>
>>> -----------------------------------------------------------------------
>>> The AMBER Mail Reflector
>>> To post, send mail to amber_at_scripps.edu
>>> To unsubscribe, send "unsubscribe amber" (in the *body* of the email)
>>> to majordomo_at_scripps.edu
>>>
>>>
>>
>>
>>
>> --
>> Wang,Ying
>>
>> -----------------------------------------------------------------------
>> The AMBER Mail Reflector
>> To post, send mail to amber_at_scripps.edu
>> To unsubscribe, send "unsubscribe amber" (in the *body* of the email)
>> to majordomo_at_scripps.edu
>>
>
> -----------------------------------------------------------------------
> The AMBER Mail Reflector
> To post, send mail to amber_at_scripps.edu
> To unsubscribe, send "unsubscribe amber" (in the *body* of the email)
> to majordomo_at_scripps.edu
>
-----------------------------------------------------------------------
The AMBER Mail Reflector
To post, send mail to amber_at_scripps.edu
To unsubscribe, send "unsubscribe amber" (in the *body* of the email)
to majordomo_at_scripps.edu
- Next message: Wang,Ying: "Re: AMBER: Non bond list error"
- Previous message: Gustavo Seabra: "Re: AMBER: pdb structure is only half the molecule."
- In reply to: Robert Duke: "Re: AMBER: Non bond list error"
- Next in thread: neville forlemu: "Re: AMBER: Non bond list error"
- Reply: neville forlemu: "Re: AMBER: Non bond list error"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
|
VU Home |
VUMC Home |
People Finder |
University Calendar
webmaster- modified on January 30, 2009 |