 | |||||||||||||||
 |
 |
 |
|
 |
|
 |
|
 |
|
 |
|
 |
|
 |
 |
AMBER Archive (2008)
Subject: Re: AMBER: interaction energies in Amber
From: Alessandro Nascimento (al.s.nascimento_at_gmail.com)
Date: Mon Jul 28 2008 - 19:51:01 CDT
- Next message: Dong Xu: "AMBER: Energy minimization of protein-ligand complex"
- Previous message: Bill Ross: "Re: AMBER: Charge neutralization, GlcNac & GalNac improper torsional angles from old PREP"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
I sent this e-mail before but didn't get it... maybe a problem with my
account...
****
Thanks, David, Carlos and Alan.
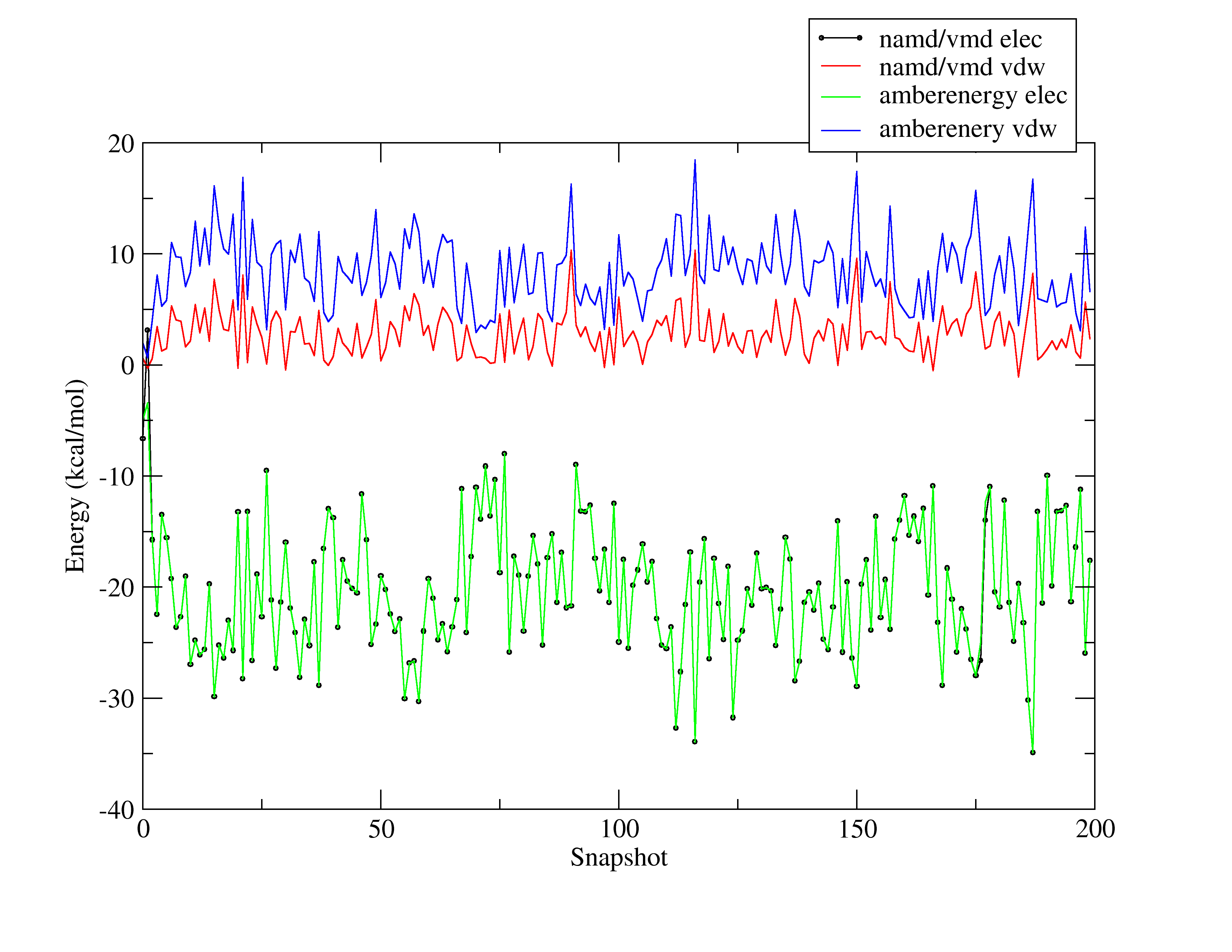
I am attaching a picture showing how different the results are.
These results were from a simulation of a waterbox (TIP3P) using
sander. I choose a waterbox to debug the contribution of the 1-4vdw
term (which is missing so far in my simple code).
The system was setup using leap as follows:
wat = createunit wat
solvatebox water TIP3PBOX 12.0
Then I minimized the system in sander, and ran a productive run
(100000 steps) also in sander, turning ntb=0.
So far, I ve being defining vdw as :
iaci = Natomtypes*(iac[i]-1);
ic = ico[(iaci+iac[j])-1]; // the `-1` corrects
for the array index which start in 0 rather than 1
if (ic > 0) {
vdw = vdw + (((LJA[ic-1])/(r2*r2*r2*r2*r2*r2))-((LJB[ic])/(r2*r2*r2))); }
else {
vdw = vdw +
(((LJA[ic-1])/(r2*r2*r2*r2*r2*r2))-((LJB[ic])/(r2*r2*r2*r2*r2))); }
Using vmd to compute the interaction energy between molecule 1 and the
other ones (using the parameters suggested in the namd manual for
amber ff simulations:
switching off
exclude 1-4scaling
1-4scaling 0.83333
cutoff 35.0
gives the results I show you in the picture
Any idea of what I am missing?
Thanks a lot,
--alessandro
- image/png attachment: Untitled.png
{kind=link}
The AMBER Mail Reflector
To post, send mail to amber_at_scripps.edu
To unsubscribe, send "unsubscribe amber" (in the *body* of the email)
to majordomo_at_scripps.edu
- Next message: Dong Xu: "AMBER: Energy minimization of protein-ligand complex"
- Previous message: Bill Ross: "Re: AMBER: Charge neutralization, GlcNac & GalNac improper torsional angles from old PREP"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
|
VU Home |
VUMC Home |
People Finder |
University Calendar
webmaster- modified on January 30, 2009 |