 | |||||||||||||||
 |
 |
 |
|
 |
|
 |
|
 |
|
 |
|
 |
|
 |
 |
AMBER Archive (2008)
Subject: Re: AMBER: interaction energies in Amber and VMD/NAMD
From: Alessandro Nascimento (al.s.nascimento_at_gmail.com)
Date: Sun Jul 27 2008 - 19:38:56 CDT
- Next message: Hannes Wallnoefer: "AMBER: radius of calcium ion in MM_PBSA"
- Previous message: Ross Walker: "RE: AMBER: big molecule numbers ( simulation with many formamide molecules )"
- In reply to: Alan: "Re: AMBER: interaction energies in Amber and VMD/NAMD"
- Next in thread: Alan: "Re: AMBER: interaction energies in Amber and VMD/NAMD"
- Reply: Alan: "Re: AMBER: interaction energies in Amber and VMD/NAMD"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
Thanks, David, Carlos and Alan.
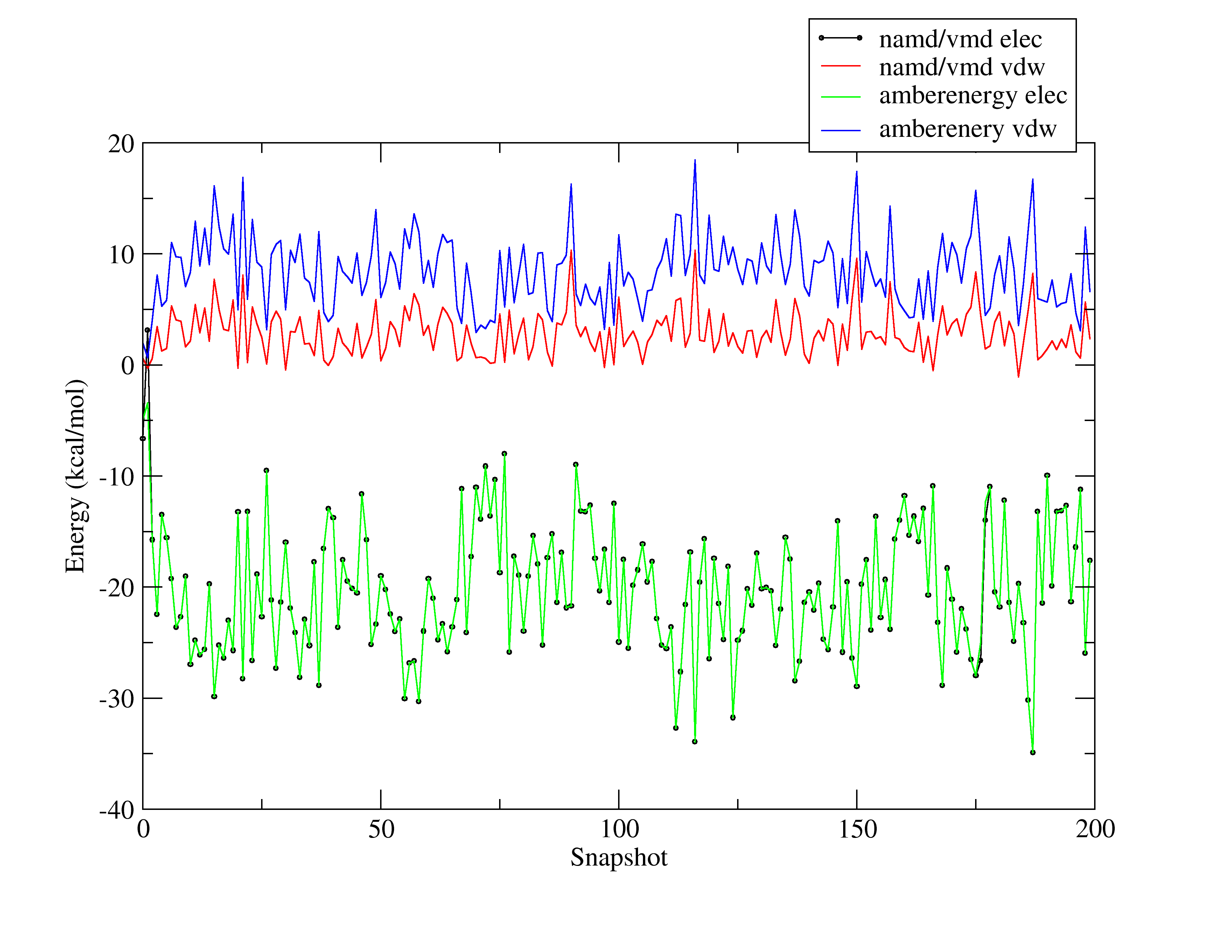
I am attaching a picture showing how different the results are.
These results were from a simulation of a waterbox (TIP3P) using
sander. I choose a waterbox to debug the contribution of the 1-4vdw
term (which is missing so far in my simple code).
The system was setup using leap as follows:
wat = createunit wat
solvatebox water TIP3PBOX 12.0
Then I minimized the system in sander, and ran a productive run
(100000 steps) also in sander, turning ntb=0.
So far, I ve being defining vdw as :
iaci = Natomtypes*(iac[i]-1);
ic = ico[(iaci+iac[j])-1]; // the `-1` corrects
for the array index which start in 0 rather than 1
if (ic > 0) {
vdw = vdw + (((LJA[ic-1])/(r2*r2*r2*r2*r2*r2))-((LJB[ic])/(r2*r2*r2))); }
else {
vdw = vdw +
(((LJA[ic-1])/(r2*r2*r2*r2*r2*r2))-((LJB[ic])/(r2*r2*r2*r2*r2))); }
Using vmd to compute the interaction energy between molecule 1 and the
other ones (using the parameters suggested in the namd manual for
amber ff simulations:
switching off
exclude 1-4scaling
1-4scaling 0.83333
cutoff 35.0
gives the results I show you in the picture
Any idea of what I may be missing?
Thanks a lot,
--alessandro
On Sat, Jul 26, 2008 at 11:26 PM, Alan <alanwilter_at_gmail.com> wrote:
> Alessandro,
>
> Have you looked at http://www.ks.uiuc.edu/Research/namd/2.6/ug/node15.html?
>
> Pay well attention to:
>
> 3. NAMD has several exclusion policy options, defined by exclude. The
> way AMBER dealing with exclusions corresponds to the ``scaled1-4'' in
> NAMD. So for simulations using AMBER force field, one would specify
> ``exclude scaled1-4'' in the configuration file, and set 1-4scaling to
> the inverse value of SCEE as would be used in AMBER.
>
> 5. By default, NAMD applies switching functions to the non-bond
> interactions within the cutoff distance, which helps to improve energy
> conservation, while AMBER does not use switching functions so it
> simply truncates the interactions at cutoff. However, if ``authentic''
> AMBER cutoff simulations are desired, the switching functions could be
> turned off by specifying ``switching off'' in NAMD configuration file.
>
> and check those parameters in your namd.conf file:
> 1-4scaling 0.833333 # =1/1.2, default is 1.0
> scnb 2 # This is default
> switching off # Turn off the switching functions, try on also
> readexclusions yes #you may compare with 'no' too.
>
> And , as David ask, how much is the difference in percentage?
>
> Cheers,
> Alan
>
> On Sun, Jul 27, 2008 at 1:46 AM, Carlos Simmerling
> <carlos.simmerling_at_gmail.com> wrote:
>> I don't think Alessandro used Amber for energies, and if it's a simple program
>> it probably is non-periodic. I think it's likely the 1-4 scaling factor for vdw.
>> one of them ( can't recall, Adrian may know) either ele or vdw has the
>> wrong 1-4 scaling in NAMD for amber ff.
>>
>> On Sat, Jul 26, 2008 at 8:36 PM, David A. Case <case_at_scripps.edu> wrote:
>>> On Sat, Jul 26, 2008, Alessandro Nascimento wrote:
>>>>
>>>> I wrote a simple program to read some information in the prmtop file
>>>> and compute nonbonded energies (ele+vdw) from a sander/pmemd
>>>> trajectory and I am trying now to compare my first test results with
>>>> the ones from the plugin NAMDEnergy in VMD. For electrostatic
>>>> computation, they match perfectly. For vdw, however, seems like
>>>> vmd/namd overestimates the energetic contribution. Does anyone has any
>>>> experience on comparing results between amber and vmd/namd ?
>>>
>>> I don't have experience, but as a guess: Amber by default includes a
>>> correction for vdw interactions beyond the cutoff; NAMD might not inlcude
>>> such a term. How different are the results?
>>>
>>> ...dac
>>>
>>> -----------------------------------------------------------------------
>>> The AMBER Mail Reflector
>>> To post, send mail to amber_at_scripps.edu
>>> To unsubscribe, send "unsubscribe amber" (in the *body* of the email)
>>> to majordomo_at_scripps.edu
>>>
>>
>>
>>
>> --
>> ===================================================================
>> Carlos L. Simmerling, Ph.D.
>> Associate Professor Phone: (631) 632-1336
>> Center for Structural Biology Fax: (631) 632-1555
>> CMM Bldg, Room G80
>> Stony Brook University E-mail: carlos.simmerling_at_gmail.com
>> Stony Brook, NY 11794-5115 Web: http://comp.chem.sunysb.edu
>> ===================================================================
>> -----------------------------------------------------------------------
>> The AMBER Mail Reflector
>> To post, send mail to amber_at_scripps.edu
>> To unsubscribe, send "unsubscribe amber" (in the *body* of the email)
>> to majordomo_at_scripps.edu
>>
>
>
>
> --
> Alan Wilter S. da Silva, D.Sc. - CCPN Research Associate
> Department of Biochemistry, University of Cambridge.
> 80 Tennis Court Road, Cambridge CB2 1GA, UK.
>>>http://www.bio.cam.ac.uk/~awd28<<
> -----------------------------------------------------------------------
> The AMBER Mail Reflector
> To post, send mail to amber_at_scripps.edu
> To unsubscribe, send "unsubscribe amber" (in the *body* of the email)
> to majordomo_at_scripps.edu
>
-- [ ]s--alessandro
- image/png attachment: Untitled.png
{kind=link}
- Next message: Hannes Wallnoefer: "AMBER: radius of calcium ion in MM_PBSA"
- Previous message: Ross Walker: "RE: AMBER: big molecule numbers ( simulation with many formamide molecules )"
- In reply to: Alan: "Re: AMBER: interaction energies in Amber and VMD/NAMD"
- Next in thread: Alan: "Re: AMBER: interaction energies in Amber and VMD/NAMD"
- Reply: Alan: "Re: AMBER: interaction energies in Amber and VMD/NAMD"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
|
VU Home |
VUMC Home |
People Finder |
University Calendar
webmaster- modified on January 30, 2009 |