 | |||||||||||||||
 |
 |
 |
|
 |
|
 |
|
 |
|
 |
|
 |
|
 |
 |
AMBER Archive (2008)
Subject: Re: AMBER: hybrid remd imaging
From: Carlos Simmerling (carlos.simmerling_at_gmail.com)
Date: Mon Mar 24 2008 - 10:59:24 CDT
- Next message: Geoff Wood: "Re: AMBER: hybrid remd imaging"
- Previous message: Geoff Wood: "AMBER: hybrid remd imaging"
- In reply to: Geoff Wood: "AMBER: hybrid remd imaging"
- Next in thread: Geoff Wood: "Re: AMBER: hybrid remd imaging"
- Reply: Geoff Wood: "Re: AMBER: hybrid remd imaging"
- Reply: Geoff Wood: "Re: AMBER: hybrid remd imaging"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
you're right, that doesn't seem to be working correctly.
can you send me directly your sander input? also do you have
anything else in the system except the 1 peptide and water?
On Mon, Mar 24, 2008 at 11:51 AM, Geoff Wood <geoffrey.wood_at_epfl.ch> wrote:
> Dear Amber Community,
> I am curious about the imaging done in hybrid remd calculations.
>
> If I use ptraj commands (see below) to post process a trajectory then the
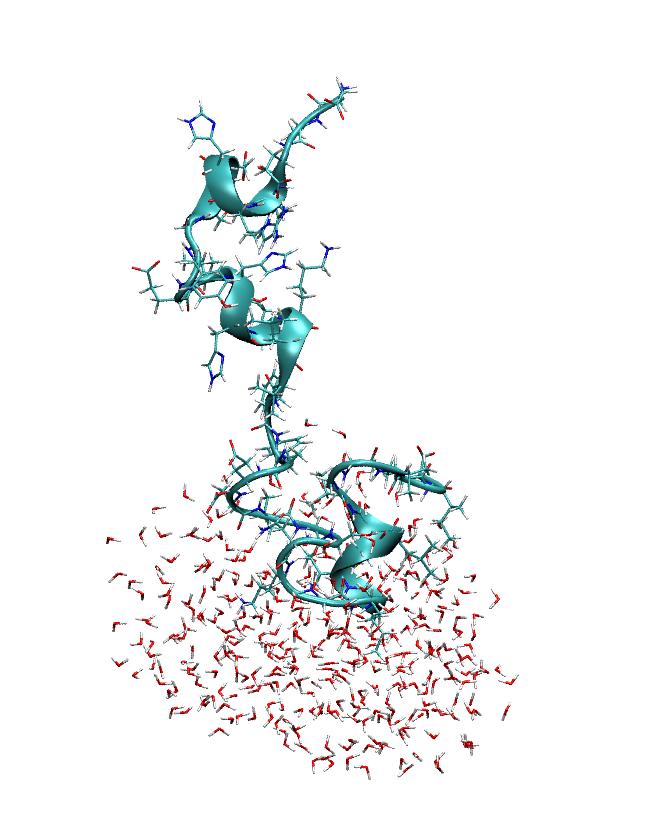
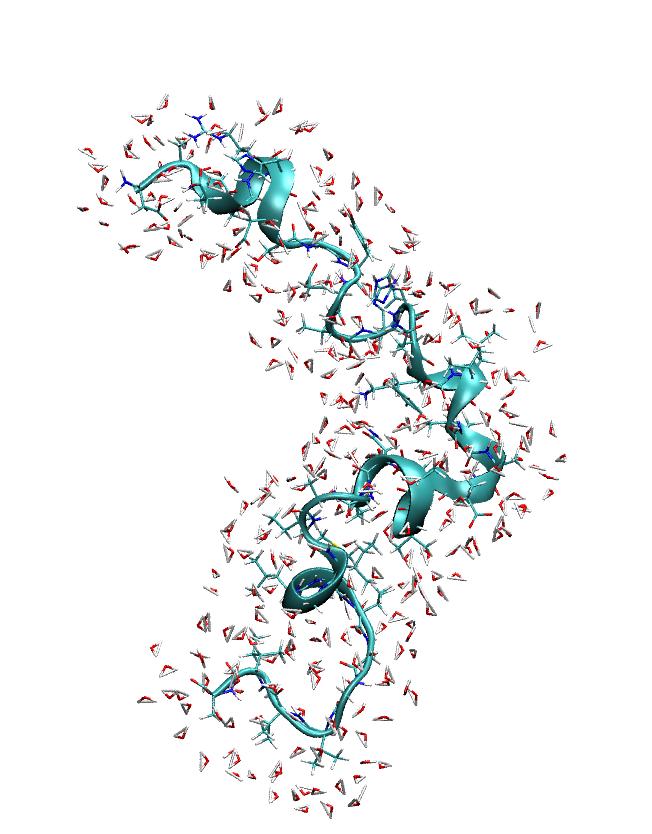
> imaged trajectory looks like what one would expect (see attached picture).
> However, if I look at the stripped restart file then the imaging seems to
> be a bit strange (see the other attached picture). Could anyone comment on
> this?
>
>
> ptraj commands:
>
> *trajin coords*
> *center * mass origin*
> *image origin center*
> *solvent byname WAT TIP3*
> *closest 350 :mask first*
> *trajout coords.strip nobox*
>
> Thanks,
>
> Dr Geoffrey Wood
> Ecole Polytechnique Fédérale de Lausanne
> SB - ISIC - LCBC
> BCH 4108
> CH - 1015 Lausanne
>
>
>
>
-- =================================================================== Carlos L. Simmerling, Ph.D. Associate Professor Phone: (631) 632-1336 Center for Structural Biology Fax: (631) 632-1555 CMM Bldg, Room G80 Stony Brook University E-mail: carlos.simmerling_at_gmail.com Stony Brook, NY 11794-5115 Web: http://comp.chem.sunysb.edu ===================================================================
 -----------------------------------------------------------------------
The AMBER Mail Reflector
To post, send mail to amber_at_scripps.edu
To unsubscribe, send "unsubscribe amber" to majordomo_at_scripps.edu
-----------------------------------------------------------------------
The AMBER Mail Reflector
To post, send mail to amber_at_scripps.edu
To unsubscribe, send "unsubscribe amber" to majordomo_at_scripps.edu
- Next message: Geoff Wood: "Re: AMBER: hybrid remd imaging"
- Previous message: Geoff Wood: "AMBER: hybrid remd imaging"
- In reply to: Geoff Wood: "AMBER: hybrid remd imaging"
- Next in thread: Geoff Wood: "Re: AMBER: hybrid remd imaging"
- Reply: Geoff Wood: "Re: AMBER: hybrid remd imaging"
- Reply: Geoff Wood: "Re: AMBER: hybrid remd imaging"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
|
VU Home |
VUMC Home |
People Finder |
University Calendar
webmaster- modified on January 30, 2009 |