 | |||||||||||||||
 |
 |
 |
|
 |
|
 |
|
 |
|
 |
|
 |
|
 |
 |
AMBER Archive (2008)
Subject: RE: AMBER: QM/MM Heating
From: Ross Walker (ross_at_rosswalker.co.uk)
Date: Mon Jan 21 2008 - 10:41:31 CST
- Next message: saurabh agrawal: "AMBER: convergence of free energy calculations"
- Previous message: Adrian Roitberg: "Re: AMBER: QM/MM Heating"
- In reply to: Steven Winfield: "AMBER: QM/MM Heating"
- Next in thread: Steven Winfield: "Re: AMBER: QM/MM Heating"
- Reply: Steven Winfield: "Re: AMBER: QM/MM Heating"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
Hi Steven
Sorry to take so long to reply to this thread. I have been travelling of
late and have only just had a chance to catch up with emails on the Amber
reflector.
What you are seeing here stems from a number of quite complex issues
involving how QM/MM interactions are handled. The manual should probably be
clarified to refer to gas phase and solvent cap simulations when talking
about energy conservation. It is also possible that tight_p_conv=1 will be
required for small QM regions to conserve energy in the gas phase. This
ensures convergence on both the energy and density rather than just the
energy.
However, that said, energy will not be conserved when running periodic
boundaries (except for DFTB in Amber 10) because a cutoff is still in use.
This is true even with QM PME because the nature of the interaction inside
and outside the cutoff is different.
In a MM calculation inside the cutoff the interaction is a pairwise charge
charge interaction. Outside the cutoff it is approximated with PME which
uses a grid to give the effective potential stemming from the infinite
pairwise interactions. This is not perfect but is accurate enough that
energy is conserved over many nanoseconds. However, in the semi-empirical
QM/MM the interaction within the QM cutoff consists of a multipole
interaction. That is the MM atoms interact with the QM density directly, not
via a pairwise approximation. However, outside of the QM cutoff a PME
approximation is used. However, it is not possible (as far as I know) to do
this in the same multipole approach used inside the cutoff and so instead
the QM atom densities are reduced to mulliken charges and these are used for
the PME (which feeds back into the fock matrix to ensure self consistency).
This means that there is a disconnect at the boundary of the QM cutoff. So
while the energies and forces are accurate for a given snapshot, the act of
a molecule leaving the QM cutoff region, translating and then re-entering
the QM cutoff region is not conservative and will lead to a net heating as
you see. The effect of this is considerably magnified for a small QM region
and even more so in the case of a single water molecule since the number of
movements in and out of the cutoff is large compared to say a QM region that
is buried in a more rigid protein active site. Thus one has to run QM/MM
with periodic boundaries in either NPT or NVT although you should be able to
use a fairly weak thermostat coupling.
One approach that could be taken to address the above issue would be to use
mulliken charges (or CM3 or equivalent) inside the QM cut off such that it
matched the approach used in the PME. This would then conserve energy for a
periodic calculation. In fact this is what DFTB does which leads to much
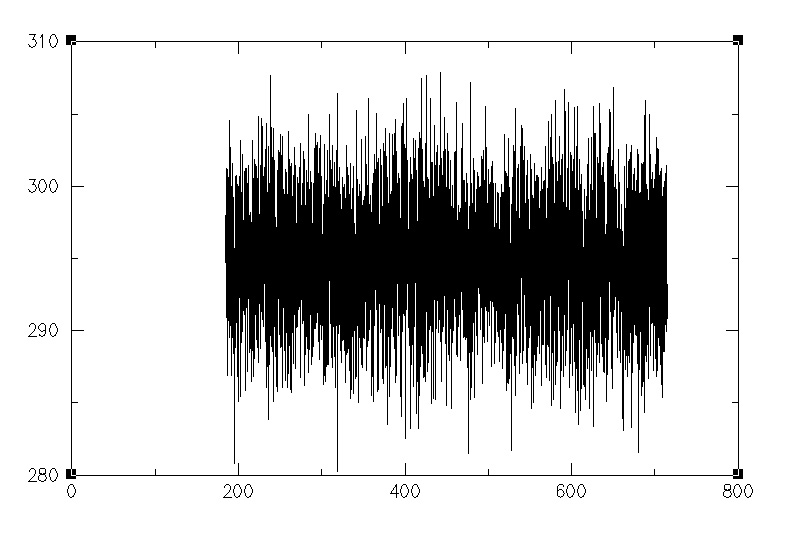
better energy conservation. See the attached plot for DFTB using your system
(although note that the density of your original prmtop and restrt file was
not equilibrated (ca. 0.9 g/cm3) so I did a much longer NPT equilibration
before switching to NVE). Note 2, however that I had to use the development
version of amber 10 to do this calculation since amber9 does not support PME
with DFTB. In theory this mulliken charge interaction could be used with the
semi-empirical approach, assuming the interaction is correctly included in
the Fock matrix to ensure self consistency. I believe that this is what
Jorgensen does in his code. However, this obviously introduces more
approximations that would require testing and validation. To be honest I
really don't know which approach is better, which one gives better radial
distribution functions etc. At present the use of muliken-muliken
interactions within the direct space cut off is not present in the Amber 9
code although I may add it at some point as an option to a later version.
Another approach would obviously be some kind of switching function at the
qm cutoff although I think the jury is still out on what artifacts such
approaches actually introduce into a simulation. For example do the
simulations then just conserve energy for the wrong reasons? I have
certainly heard discussions to the effect that such switching approaches,
just for classical simulations, can be worse than no switch at all.
So I hope that, somewhat answers your questions. One key thing to remember
is that MD simulations will only conserve energy for equilibrated systems
which yours was not, the density was way too low so this may well have
contributed to the problems you see. Plus I think you might see better
results with a more rigid system, such as a ligand in an active site rather
than highly mobile water. You could also try setting tight_p_conv=1 and
setting a larger qmcut - say 14.0 (for a larger system obviously) and this
may improve things. However, for production simulations at the moment I
would recommend running a full NVE heating, NPT equilibration (langevin) and
then switch to NPT with a weak barostat and thermostat, say taup=10.0,
tautp=10.0. For most protein systems this should be reasonable. Although as
with any scientific code/algorithm the QM/MM implementation is in need of
much more testing so I would encourage you to try things out and let me know
how they go.
All the best
Ross
/\
\/
|\oss Walker
| Assistant Research Professor |
| San Diego Supercomputer Center |
| Tel: +1 858 822 0854 | EMail:- ross_at_rosswalker.co.uk |
| http://www.rosswalker.co.uk | PGP Key available on request |
Note: Electronic Mail is not secure, has no guarantee of delivery, may not
be read every day, and should not be used for urgent or sensitive issues.
> -----Original Message-----
> From: owner-amber_at_scripps.edu
> [mailto:owner-amber_at_scripps.edu] On Behalf Of Steven Winfield
> Sent: Monday, January 14, 2008 07:22
> To: amber_at_scripps.edu
> Subject: AMBER: QM/MM Heating
>
> Dear all,
>
> I'm having a look at AMBER 9's energy conservation when using
> its internal
> QM/MM code. I took a simple system of pure water (1222
> molecules in total
> in a periodic box of length 34.419A), with one water molecule
> flagged as the QM region (using the PM3 Hamiltonian), turned
> off SHAKE for
> this molecule and used a 0.2fs time step. The other molecules
> were TIP3P.
> The input restart file had been equilibrated at 300K
> using 100ps of NVT dynamics with the Langevin thermostat. The
> QM/MM simulation lasted 1ns in total.
>
> I saved the position and velocity data so that I could examine the
> temperature of the MM and QM regions afterwards. I have
> attached a bzipped
> tar with my input files, an output text file, and some
> temperature plots.
> Measuring the temperature of a single molecule is obviously
> very noisy, so
> I also calculated a weighted average of temperature values at
> each point
> with weights exponentially decaying with a time constant of 50ps.
>
> As can be seen from the plot (pme_temp_50.ps) and the text
> output file, I
> observed a steady temperature increase from 300K to around
> 500K, and the
> average temperature of the quantum water becomes quite large.
> I tried the
> same simulation but using a full Ewald sum (ewald_temp_50.ps)
> and observed
> similar results.
>
> I ran a pure MM simulation with the same input files for
> comparison (with
> SHAKE on all molecules),
> because page 143 of the manual says that QM/MM with default
> parameters should "generally conserve energy about as well as
> one would
> find for a corresponding pure MM simulation". I increased the
> time step by
> a factor of 10 to 2.0fs, which I thought would be on the very edge of
> becoming unstable, but the temperature plot (water_mm.ps)
> shows no signs
> of heating.
>
> So my questions are:
>
> What non-default parameters have I used, if any?
> I've had others look at my input files and the only comment
> I've had is
> that my density may be slightly low. I've kept my timestep
> small and my
> cutoffs large (9.0A)
> Is there a problem with AMBER's QM/MM code? I'm using a fully patched
> version.
>
> Any comments would be most helpful.
>
> Regards,
> Steven Winfield.
>
> P.S. The input restart file is called 'zeroed' because I
> wanted initially
> to see if energy was being successfully transferred from the rigid MM
> molecules to the non-rigid bonds of the QM molecule, so I
> manually zeroed
> the velocities of the QM molecule before starting. This
> shouldn't affect
> the results though.
-----------------------------------------------------------------------
The AMBER Mail Reflector
To post, send mail to amber_at_scripps.edu
To unsubscribe, send "unsubscribe amber" to majordomo_at_scripps.edu
- Next message: saurabh agrawal: "AMBER: convergence of free energy calculations"
- Previous message: Adrian Roitberg: "Re: AMBER: QM/MM Heating"
- In reply to: Steven Winfield: "AMBER: QM/MM Heating"
- Next in thread: Steven Winfield: "Re: AMBER: QM/MM Heating"
- Reply: Steven Winfield: "Re: AMBER: QM/MM Heating"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
|
VU Home |
VUMC Home |
People Finder |
University Calendar
webmaster- modified on January 30, 2009 |