 | |||||||||||||||
 |
 |
 |
|
 |
|
 |
|
 |
|
 |
|
 |
|
 |
 |
AMBER Archive (2007)
Subject: Re: AMBER: Problem with xLeap
From: alfredoq_at_mail.fcq.unc.edu.ar
Date: Tue Nov 13 2007 - 11:07:40 CST
- Next message: David A. Case: "Re: AMBER: Problem with xLeap"
- Previous message: ABEL Stephane 984007: "AMBER: 2 double bond in AMBER ffield"
- In reply to: David A. Case: "Re: AMBER: Problem with xLeap"
- Next in thread: David A. Case: "Re: AMBER: Problem with xLeap"
- Reply: David A. Case: "Re: AMBER: Problem with xLeap"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
Dear Dr. Case: I start xleap with the command "xleap -s -f leaprc.ff99"
the following appears in the console window:
[general_at_localhost ~]$ xleap -s -f leaprc.ff99
-I: Adding /home/general/Programas/amber9/dat/leap/prep to search path.
-I: Adding /home/general/Programas/amber9/dat/leap/lib to search path.
-I: Adding /home/general/Programas/amber9/dat/leap/parm to search path.
-I: Adding /home/general/Programas/amber9/dat/leap/cmd to search path.
-s: Ignoring startup file: leaprc
-f: Source leaprc.ff99.
Warning: Cannot convert string "-*-helvetica-medium-r-*-*-18-*-*-*-*-*-*-*" to
type FontStruct
Warning: Cannot convert string "-*-helvetica-bold-r-*-14-*" to type FontStruct
Warning: Cannot convert string "-*-helvetica-medium-o-*-*-14-*-*-*-*-*-*-*" to
type FontStruct
Warning: Cannot convert string "-*-courier-bold-r-*-*-14-*-*-*-*-*-*-*"
to type
FontStruct
Warning: Cannot convert string "-*-helvetica-medium-o-*-*-18-*-*-*-*-*-*-*" to
type FontStruct
Warning: Cannot convert string "-*-courier-medium-r-*-*-14-*-*-*-*-*-*-*" to
type FontStruct
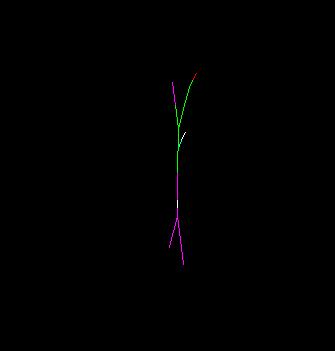
I can rotate the molecule, but only in two planes. I see a two dimensional
(flat) view of the molecule, for example the ALA residue. I am attaching the
image.
Thank you very much
Alfredo Quevedo
Quoting "David A. Case" <case_at_scripps.edu>:
> On Tue, Nov 13, 2007, alfredoq_at_mail.fcq.unc.edu.ar wrote:
>
>> I am writting in order to ask for some hint about a
>> problem with xLeap. The point is that when I load a force field, for example
>> ff99, and after trying to edit any residue, I see the molecule in a 2D plain
>> view.
>
> I'm not sure I can tell what you mean by "2D plain view". Can you give the
> exact commands you used for xleap, and a better description of what
> appears on
> the screen? Can you rotate the molecule around using the mouse? Do the bond
> lengths and angles look correct?
>
> ...dac
>
> -----------------------------------------------------------------------
> The AMBER Mail Reflector
> To post, send mail to amber_at_scripps.edu
> To unsubscribe, send "unsubscribe amber" to majordomo_at_scripps.edu
>
----------------------------------------------------------------
Facultad de Ciencias QuÃmicas - Universidad Nacional de CÃrdoba
-----------------------------------------------------------------------
The AMBER Mail Reflector
To post, send mail to amber_at_scripps.edu
To unsubscribe, send "unsubscribe amber" to majordomo_at_scripps.edu
- Next message: David A. Case: "Re: AMBER: Problem with xLeap"
- Previous message: ABEL Stephane 984007: "AMBER: 2 double bond in AMBER ffield"
- In reply to: David A. Case: "Re: AMBER: Problem with xLeap"
- Next in thread: David A. Case: "Re: AMBER: Problem with xLeap"
- Reply: David A. Case: "Re: AMBER: Problem with xLeap"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
|
VU Home |
VUMC Home |
People Finder |
University Calendar
webmaster- modified on January 30, 2009 |