 | |||||||||||||||
 |
 |
 |
|
 |
|
 |
|
 |
|
 |
|
 |
|
 |
 |
AMBER Archive (2007)
Subject: Re: AMBER: Problem related simulation of dimer
From: Sandeep Kaushik (sandy.thesmitten_at_gmail.com)
Date: Mon Mar 19 2007 - 05:16:39 CST
- Next message: Cenk Andac: "Re: AMBER: Essential ions and water molecules in MMPBSA computations"
- Previous message: priya priya: "Re: AMBER: Problem related simulation of dimer"
- In reply to: priya priya: "Re: AMBER: Problem related simulation of dimer"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
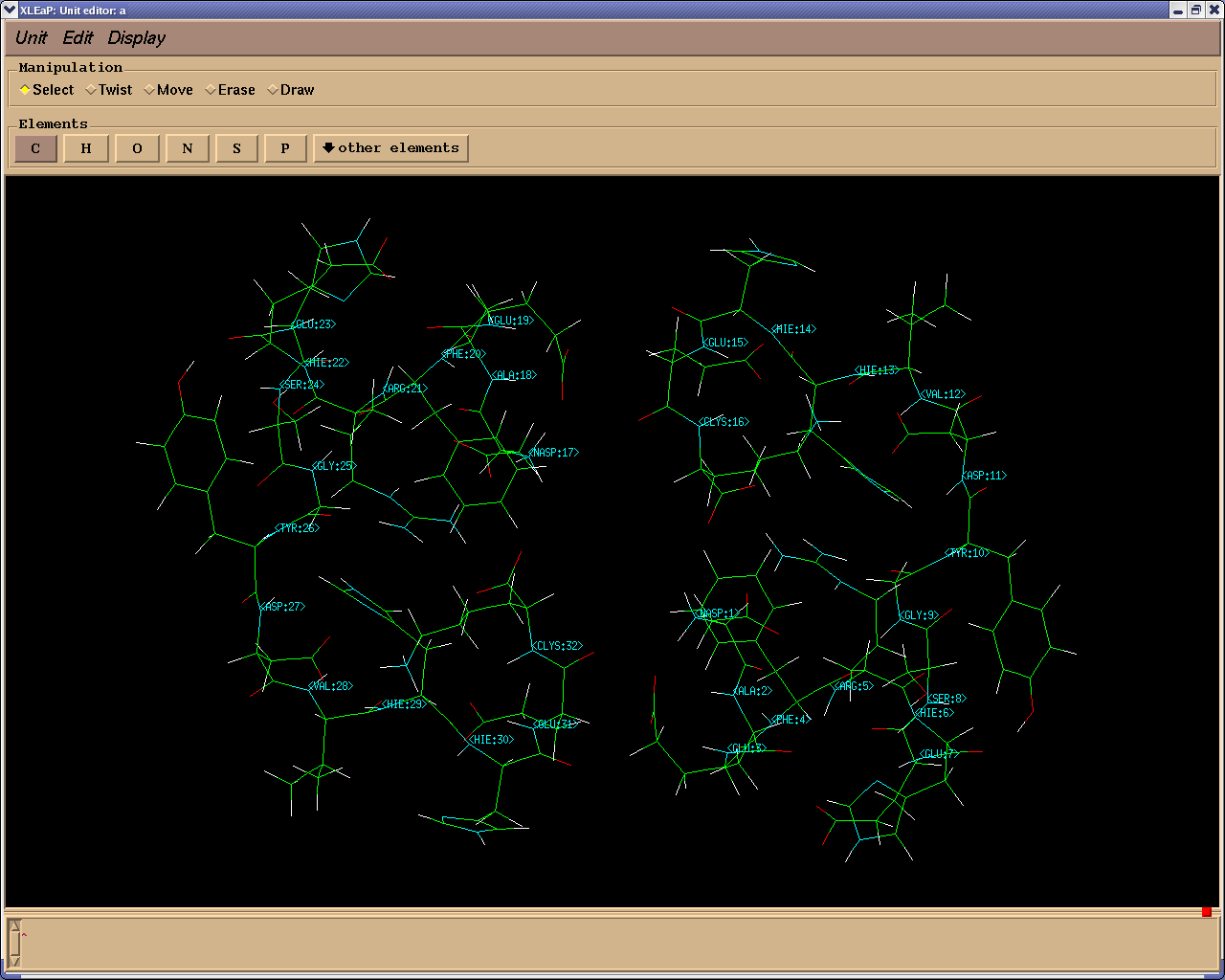
Use the attached PDB file to load the DImer..
See the attached picture (PNG file) to see how it looks after loading this
PDB file in LEaP and using EDIT command.....
Cheers.
On 3/19/07, priya priya <priyaanand_27_at_yahoo.co.in> wrote:
>
> i had already added the TER line inbetween the two chains but still it
> shows a bond.
>
> *Sandeep Kaushik <sandy.thesmitten_at_gmail.com>* wrote:
>
> Hi Priya
> You needed to add a TER card between the two chains in order to tell the
> program to know that one chains "TERminates" here and following coordinates
> belong to a new chain.. so once you do this there would be no bond forming
> b/w residue 16 & 17.... however there still would be two atoms added to your
> pdb file:
>
> Added missing heavy atom: .R<CLYS 16>.A<OXT 23>
> Added missing heavy atom: .R<CLYS 32>.A<OXT 23>
>
> This addition of the C-terminal residue Oxygen is b'coz there is no oxygen
> in the LYSINE unit of "parm99". In order to complete the terminal residue,
> the program needs to add these oxygens.. So that's logical you see..
>
> I hope this would have answered your questions..
>
> Cheers and
> Good Luck.
>
>
>
> On 3/19/07, priya priya < priyaanand_27_at_yahoo.co.in> wrote:
> >
> > hi
> > I am trying dimer's pdb file with different co-ordinates for both the
> > chains.
> > when i load this pdb file in xleap it creates a new atom named H within
> > the first residue in the second chain, and it also added a missing atom in
> > last residue of the second chain.
> > thus file contained 3 atoms not in residue templates.
> > and when i use the command edit in xleap it shows a dimer with a long
> > bond formed between the first chain C-terminal and second chain N-terminal.
> > Attaching pdb file with mail.
> >
> > Priya
> >
> > *Sandeep Kaushik < sandy.thesmitten_at_gmail.com>* wrote:
> >
> > Dear Priya,
> > I saw your dimer's PDB file. But i guess you don't have a dimer.. the
> > PDB file you have given has same coordinates for both peptide chains...
> > And the bond thing that you saw using edit in LEaP is I GUESS you are
> > talking about another mistake in the pdb file where a space has come in
> > front of the coordinate values in line number 199-201 (residue 18) so PDB
> > file is read as
> >
> > ATOM 193 CA ALA 18 2.039 2.062 1.486 1.00 0.00
> > ATOM 194 CB ALA 18 2.016 1.914 1.453 1.00 0.00
> > ATOM 195 C ALA 18 2.168 2.084 1.565 1.00 0.00
> >
> > instead of
> >
> > ATOM 193 CA ALA 18 20.392 20.621 14.865 1.00 0.00
> > ATOM 194 CB ALA 18 20.160 19.144 14.539 1.00 0.00
> > ATOM 195 C ALA 18 21.681 20.840 15.659 1.00
> > 0.00
> >
> > So i hope you need to have different coordinates to get the chains
> > differently... there would't be any problem from the TER card..
> > Cheers
> >
> >
> > On 3/15/07, priya priya <priyaanand_27_at_yahoo.co.in > wrote:
> > >
> > > Dear All
> > > I am trying to run a simulation of dimer each chain has 16 residues.Inthe pdb fileI added TER between the chains but when i use the command edit
> > > in xleap there is a bond between the chains. but i want two different chains
> > > and non bonded. I am attaching the pdb file with mail.
> > > will anybody tell me the mistake.
> > > Thanks in advance
> > > Priya
> > >
> > > ------------------------------
> > > Here's a new way to find what you're looking for - Yahoo! Answers
> > > <http://us.rd.yahoo.com/mail/in/yanswers/*http://in.answers.yahoo.com/>--0-981625490-1173957811=:66316--
> > >
> > >
> > >
> >
> >
> > --
> > sandy_thesmitten
> >
> >
> > ------------------------------
> > Here's a new way to find what you're looking for - Yahoo! Answers
> > <http://us.rd.yahoo.com/mail/in/yanswers/*http://in.answers.yahoo.com/>
> >
> >
>
>
> --
> sandy_thesmitten
>
>
> ------------------------------
> Here's a new way to find what you're looking for - Yahoo! Answers<http://us.rd.yahoo.com/mail/in/yanswers/*http://in.answers.yahoo.com/>
>
>
-- sandy_thesmitten
- image/png attachment: ABM1.png
{kind=link}
- chemical/x-pdb attachment: ABM.pdb
- Next message: Cenk Andac: "Re: AMBER: Essential ions and water molecules in MMPBSA computations"
- Previous message: priya priya: "Re: AMBER: Problem related simulation of dimer"
- In reply to: priya priya: "Re: AMBER: Problem related simulation of dimer"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
|
VU Home |
VUMC Home |
People Finder |
University Calendar
webmaster- modified on January 30, 2009 |