 | |||||||||||||||
 |
 |
 |
|
 |
|
 |
|
 |
|
 |
|
 |
|
 |
 |
AMBER Archive (2006)
Subject: Re: AMBER: use_pme=0 problem
From: luckyang_at_gmail.com
Date: Fri May 26 2006 - 15:19:11 CDT
- Next message: Bill Ross: "Re: AMBER: use_pme=0 problem"
- Previous message: David A. Case: "Re: AMBER: Please reply"
- In reply to: David A. Case: "Re: AMBER: use_pme=0 problem"
- Next in thread: Bill Ross: "Re: AMBER: use_pme=0 problem"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
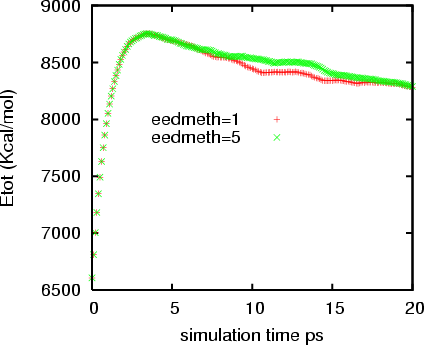
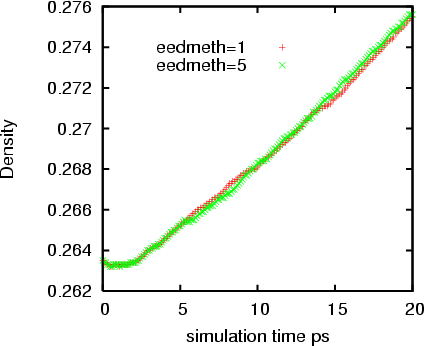
But this error accumulates as the simulation running. Although the overall
trend are still the same, I can see the difference in the potential energy
and density from a 20 ps simulation. And this difference is not trivial. I
tested a system with 3000 atoms without any partial charges. In the
attachment please find the difference of the potential energy for 20 ps
simulations with eedmeth=5 and 1.
Thanks,
Lu
On 5/26/06, David A. Case <case_at_scripps.edu> wrote:
>
> On Fri, May 26, 2006, luckyang_at_gmail.com wrote:
> >
> > 194c192
> > < Evdw = -2018.956128965737
> > ---
> > > Evdw = -2018.956128965736
>
> I think you can safely ignore differences on the order of 1 part in
> 10**16!
> Please see section 1.3.4 ("testing") in either the Amber8 or the Amber9
> manual.
>
> ....dac
>
> -----------------------------------------------------------------------
> The AMBER Mail Reflector
> To post, send mail to amber_at_scripps.edu
> To unsubscribe, send "unsubscribe amber" to majordomo_at_scripps.edu
>
 -----------------------------------------------------------------------
-----------------------------------------------------------------------
The AMBER Mail Reflector
To post, send mail to amber_at_scripps.edu
To unsubscribe, send "unsubscribe amber" to majordomo_at_scripps.edu
- Next message: Bill Ross: "Re: AMBER: use_pme=0 problem"
- Previous message: David A. Case: "Re: AMBER: Please reply"
- In reply to: David A. Case: "Re: AMBER: use_pme=0 problem"
- Next in thread: Bill Ross: "Re: AMBER: use_pme=0 problem"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
|
VU Home |
VUMC Home |
People Finder |
University Calendar
webmaster- modified on January 30, 2009 |