 | |||||||||||||||
 |
 |
 |
|
 |
|
 |
|
 |
|
 |
|
 |
|
 |
 |
AMBER Archive (2006)
Subject: AMBER: force field parameters
From: Douali, Latifa (latifa.douali_at_pnl.gov)
Date: Thu May 18 2006 - 12:33:35 CDT
- Next message: Navnit Kumar Mishra: "Re: AMBER: force field parameters"
- Previous message: David A. Case: "Re: AMBER: "The system has extended beyond" error message"
- Next in thread: Navnit Kumar Mishra: "Re: AMBER: force field parameters"
- Reply: Navnit Kumar Mishra: "Re: AMBER: force field parameters"
- Maybe reply: Douali, Latifa: "RE: AMBER: force field parameters"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
Dear AMBER users,
I am running an MD simulation for a non standard nucleic acid which is
non planar.
In this structure, I have a methyl group in a pseudo axial position.
My question is:
1) how can I prevent this methyl from inverting to the equatorial
position.
The improper dihedrals I used for these simulations are as follow:
CT-C-N*-CT 1.1 180.0 2.0
CT-C-N*-CM 1.1 180.0 2.0
C-CT-CT-CT 1.1 180.0 2.0
N*-OH-CT-HC 1.1 180.0 2.0
CT-OH-CT-HC 1.1 180.0 2.0
N*-CT-CT-HC 1.1 180.0 2.0
2) May the proper dihedral angles also play a role in this transition
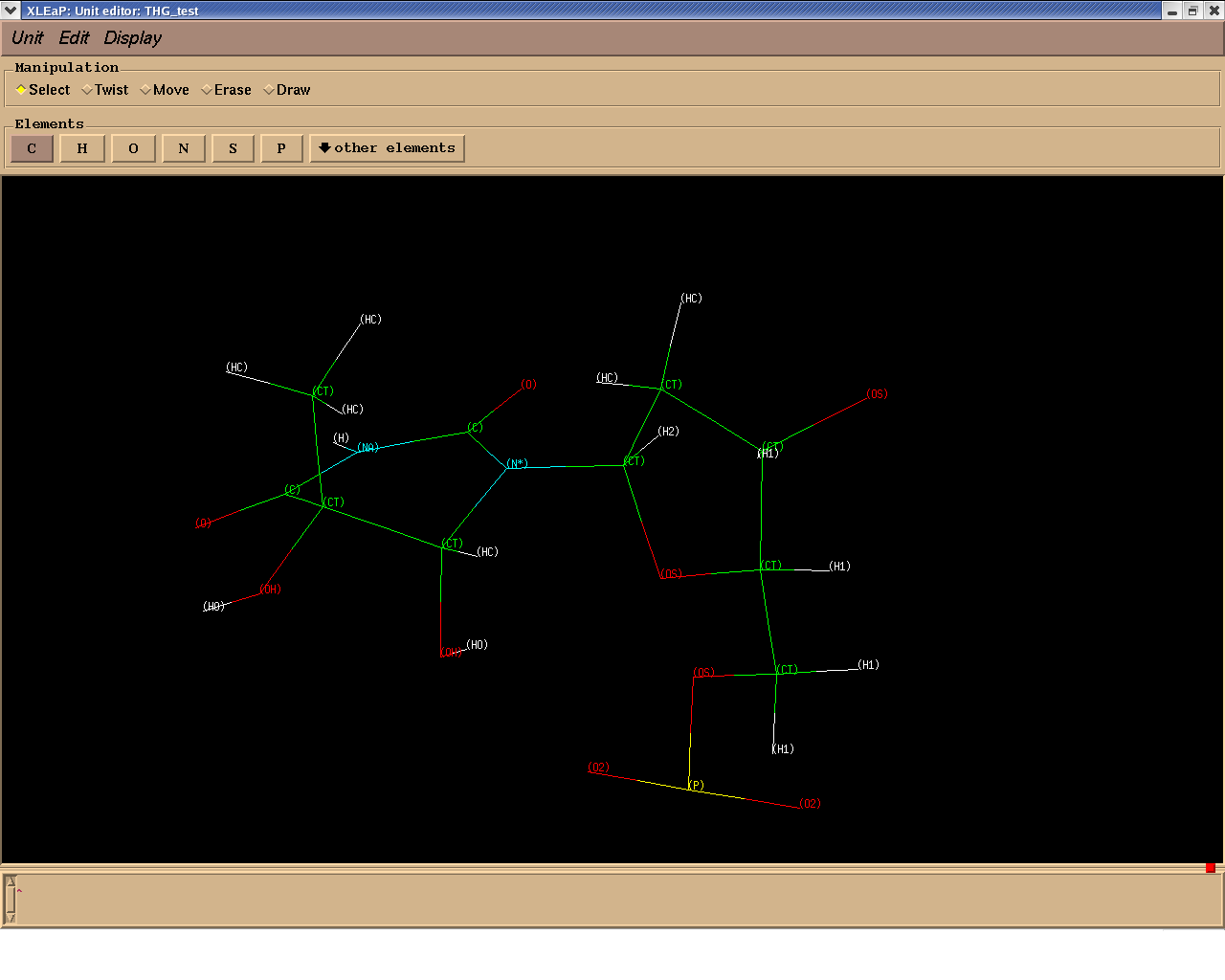
Attached is the structure of this system with the atom types assigned. I
might be doing something wrong,
any advice will be welcome
Latifa
- image/png attachment: thg_types.png
{kind=link}
The AMBER Mail Reflector
To post, send mail to amber_at_scripps.edu
To unsubscribe, send "unsubscribe amber" to majordomo_at_scripps.edu
- Next message: Navnit Kumar Mishra: "Re: AMBER: force field parameters"
- Previous message: David A. Case: "Re: AMBER: "The system has extended beyond" error message"
- Next in thread: Navnit Kumar Mishra: "Re: AMBER: force field parameters"
- Reply: Navnit Kumar Mishra: "Re: AMBER: force field parameters"
- Maybe reply: Douali, Latifa: "RE: AMBER: force field parameters"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
|
VU Home |
VUMC Home |
People Finder |
University Calendar
webmaster- modified on January 30, 2009 |