 | |||||||||||||||
 |
 |
 |
|
 |
|
 |
|
 |
|
 |
|
 |
|
 |
 |
AMBER Archive (2009)
Subject: [AMBER] HOW TO OPTIMISE INDI-DIELC_MM VALUE for MM_PBSA calculation ?
From: Marek Maly (marek.maly_at_ujep.cz)
Date: Tue Dec 22 2009 - 12:49:42 CST
- Previous message: Frank X. Vázquez: "[AMBER] Van der Waals and electrostatic Forces"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
Dear all,
I would like to ask for some suggestions regarding to optimal setting of
INDI ( Dielectric constant for the solute )
and so also DIELC parameter (@MM section) which is required to be the same
as INDI, regarding to MM/PBSA analysis.
Here are some representative ones:
http://ambermd.org/Questions/mail/212.html
http://archive.ambermd.org/200008/0041.html
http://archive.ambermd.org/200504/0282.html
http://archive.ambermd.org/200210/0086.html
I know that "solute dielectric constant" topic was already discussed. I
read lot of the contributions in Amber archive and
also some relevant articles. I learned that let say physically correct is
the default setting INDI=1 which is argumented for example like this
" The dielectric constant of all solute was uniformly set to 1.0, as in
the calibration of the force field for protein."
Lu, Q.; Luo, R. A Poisson-Boltzmann dynamics method with nonperiodic
boundary condition. J. Chem. Phys., 2003, 119, 11035-11047
or
"The only thing that is consistent with the way charges are derived is to
set intdiel=1 (the default, and recommended value)."
http://archive.ambermd.org/200504/0282.html
This argumentation was related MM/GBSA but If I am not wrong it is
probably valid also for MM/PBSA.
It is probably related to the fact that forcefield parameters are usually
derived from QM energy calculations of small molecular fragments
in vacuum. Am I right ?
Anyway sometime the default setting (INDI=1) could bring discutable
results - please see my older contributions:
http://archive.ambermd.org/200906/0197.html
http://archive.ambermd.org/200906/0255.html
So one can think about experiments with some parameters like INDI and
consequently also with DIELC (@MM section) since DIELC (@MM section)
is required to be the same as INDI. I am not sure if I understood this
requirement well since @MM section refers to explicit calculations
in vacuum (El., VDW) so only value 1 is the meaningful here but INDI is
related to the implicit PB calculations where the proper dielctric.
constant.
of the solute could be seen as some "semiempirical" parameter, which could
have recommended value 1 (the physically meaningful/consistent with @MM)
but
not necessarily equal to DIELC_at_MM. But here probably "consistency"
requirement of the same diel. constant in case of @MM and @PB won. Am I
right ?
There is also another problem if one admit el. diel. of solute (implicit
calculations) different from 1, and this could be differences for different
kinds of molecules.
Here are for completeness' sake just two examples of SW where the default
values for solute diel. c. in case of PB calculations are different from 1.
Pymol APBS Tools
-----------------
Protein Dielectric: 2.0
Solvent dielectric: 80.0
H++
--------
Internal Dielectric: 6
External Dielectric: 80
Anyway I don't want to discuss this topic on the theoretical level but
much rather from the practical one and my main question
which will be probably interesting also for the others is: HOW TO OPTIMISE
INDI/DIELC_at_MM VALUE TO OBTAIN THE MOST RELIABLE RESULTS
OF THE MM/PBSA ANALYSIS ?
The interpretation of this question is of course how to find INDI/DIELC_at_MM
to obtain dG which is the closest to the experimental one or to
that which could be theoretically achieved from very very long
trajectories using just explict calculations.
Of course in the situation (which is quite frequent), that the exper.
value. is unknown.
I am pretty aware of the fact that dG is composed of several components
which has also their parameters ..., but I would like to focus
here just on contribution of INDI/DIELC_at_MM parameters assuming that all
the other parameters are fixed.
Here are my two ideas to this topic and I would be very grateful for any
relevant comments and of course for suggestions of another approaches.
#1
I did some experiments with one of my "disobedient" complex, which reveal
a nice complexation but calculated dG was untrustworthy - please see:
http://archive.ambermd.org/200906/0197.html
http://archive.ambermd.org/200906/0255.html
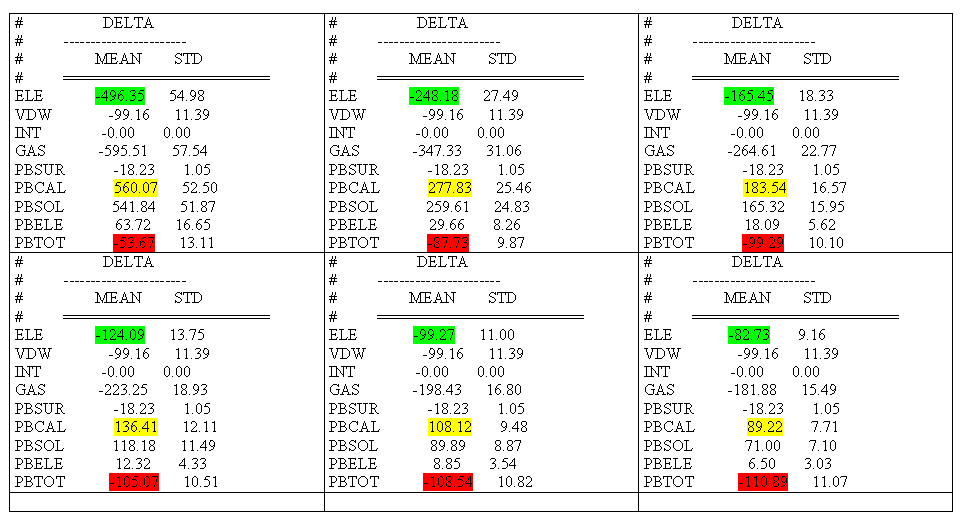
I recalculated dH (dG) of the given system for different values of
INDI/DIELC_at_MM and here are the results:
INDI & DIELC (@MM section) PBTOT dG
1 -53.67 20.86
2 -87.73 -13.20
3 -99.29 -24.76
4 -105.07 -30.54
5 -108.54 -34.01
6 -110.89 -36.36
TSTOT -74.53 ( DIELC=4 @NN section )
The details you can see in attached file with the most affected dH
contributions highlighted (order from left to right and from top to
bottom).
As you can see, there is start of saturation evident near the value
INDI/DIELC_at_MM = 6 . Could be this
a resonable fact to consider INDI/DIELC_at_MM around 6 or PBTOT around -110
as the resonable values ?
#2
There could be some indirect way through the comparison of some
characteristics (for example radial distr. function of the system of
interest or of some representative parts)
obtained from MD trajectories (explicit solvent) and MD trajectories
(implicit solvent). The idea is simple: to do several MD runs (implicit
solvent) with different pameter of interest
(here solute dielectric const.) and compare the relevant charcteristic
with that obtained from MD (explicit solvent). But I have to say that I
never did MD in implicit solvent so
I am not sure if the proposed approach is meaningful ...
Thanks to all in advance for any relevant contribution, especially for
suggesting of some own approach which was already successfully used/tested
!
Best wishes,
Marek
-- Tato zpráva byla vytvoøena pøevratným po¹tovním klientem Opery: http://www.opera.com/mail/
- image/png attachment: INDI_DIELC_effect.png
{kind=link}
_______________________________________________
AMBER mailing list
AMBER_at_ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
- Previous message: Frank X. Vázquez: "[AMBER] Van der Waals and electrostatic Forces"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
|
VU Home |
VUMC Home |
People Finder |
University Calendar
webmaster- modified on December 22, 2009 |