 | |||||||||||||||
 |
 |
 |
|
 |
|
 |
|
 |
|
 |
|
 |
|
 |
 |
AMBER Archive (2009)
Subject: [AMBER] Mixing of forcefields in case of covaletly bonded systems
From: Marek Malý (maly_at_sci.ujep.cz)
Date: Mon Apr 27 2009 - 07:06:38 CDT
- Previous message: Carlos Simmerling: "Re: [AMBER] Error with calculation of the potential energy"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
Dear all,
as it is clear for example from this discussion "Possibility to use ff99 +
GAFF + GLYCAM_06 in one time for complex system ?" there
is no problem to use several different ff (e.g. ff99SB, GAFF, GLYCAM_06)
in one time for Amber calculation of some complex system.
Situation is relatively easy if all the parts of the simulated system
which are treated using different forcefields are separated.
Situation becomes more complicated in the case when two or more parts of
the system which are treated using different ff are covalently
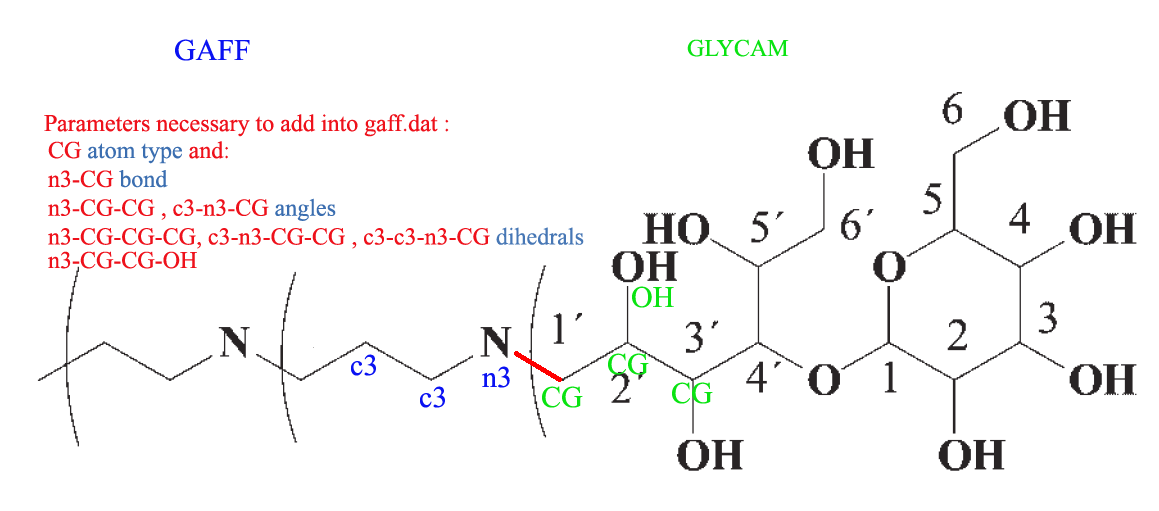
bonded. I illustrated this situation on attached picture where you can see
end of PPI dendrimer branch (treated with GAFF) with covalently bound
maltose ( for which GLYCAM_06 is recommended).
The covalent bond connecting terminal dendrimer amine and 1' maltose
carbon is highlighted in red.
Of course now we have to solve somehow ff discontinuity near that covalent
bond.
From my above mentioned discussion with Karl Kirschner is clear that one
solution is to add some "mixed" terms into
gaff ff or more precise into gaff.dat (please see the attached picture
where is written the minimal set of these parameters for the given system).
Since from several reasons could be complicated to use this approach
(e.g. no possibility to edit gaff.dat on the given computer cluster;
problems with own Amber installation in HOME dir of the given cluster ) I
would like to know if could be enough to create just proper FRCMOD file
to add this mixed terms (like for example in the case of in Amber actually
non-parametrised elements like for example silicon etc.)
Of course that this is question especially for the Amber developers but
any opinion is welcomed especially from
the Amber users who already tried to solve similar problem.
Best regards
Marek
-- Tato zpráva byla vytvoøena pøevratným po¹tovním klientem Opery: http://www.opera.com/mail/
- image/png attachment: DEN_MAL.png
{kind=link}
_______________________________________________
AMBER mailing list
AMBER_at_ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
- Previous message: Carlos Simmerling: "Re: [AMBER] Error with calculation of the potential energy"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
|
VU Home |
VUMC Home |
People Finder |
University Calendar
webmaster- modified on April 27, 2009 |