 | |||||||||||||||
 |
 |
 |
|
 |
|
 |
|
 |
|
 |
|
 |
|
 |
 |
AMBER Archive (2009)
Subject: [AMBER] FF Parameters Transformations ...?
From: Marek Malý (maly_at_sci.ujep.cz)
Date: Wed Apr 01 2009 - 12:29:26 CDT
- Previous message: Seth Hayik: "Re: [AMBER] make test.serial.QMMM"
- In reply to: Marek Malý: "[AMBER] Parametrisation of Silicon based molecules for Amber simulations ?"
- Next in thread: Karl Kirschner: "Re: [AMBER] FF Parameters Transformations ...?"
- Reply: Karl Kirschner: "Re: [AMBER] FF Parameters Transformations ...?"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
Dear Amber users,
some time ago I sent to this forum the
below attached email regarding to calculations of Si-based molecules in
Amber.
Unfortunately I didn't receive any response till now :((
Probably it is not so easy to answer/help here as it is clear to me from
what
I read about similar problems in this forum.
So I have to this story just 2 additional questiones ( hopefully not so
complicated as the original one :)) )
#1 - as it is clear antechamber/parmchk routine has problem to associate
Si atoms
with the proper bond, angle, dihedral parameters since Amber
forcefileds do not
contain this parameters for silicon so it is necessary to look for it
somewhere
elsewhere and modify properly relevant FRCMOD file. I found this
parameters in
CVFF a PCFF forcefields so I will try to transport/transfer them for
the Amber calculation. Is it a good idea ? What I should to care
about ?
Probably to check the physical units, check if mathematical
expressions for
the given type of forces is the same i both forcefields, if not make
some relevant transformation
of the transfered parameters ?
Maybe could be a good/elegant idea to use well known
parametrisations/parameters (like for example C-N, C-N-C, C-C-N-C)
which are present in CVFF, PCFF but also in Amber ff to calculate
some scaling constant/s which can be used for
easy transformation of C-Si, C-Si-C ... parameters from CVFF, PCFF
into Amber ff. Could be this a good idea ?
Another possibility is to use PARMCAL routine - I have never used it
till now so it would the case to use it to
obtain at least the bond length and angle ff parameters for C-Si,
Si-O, C-C-Si, C-Si-O ... bonds/angles ?
Any experiences with this ffx->ffy transformations ... are highly
welcomed ! I think that this issue could be a very interesting
also for the other Amber users.
#2 - On contrary it seems to me that antechamber/difcon ... has absolutely
no problems with "exotic" Si atoms
and for example calculated proper(?) Si partial charges. In PREPIN
file (please see attached file), there is
no error reported so I just would like to ensure: Is it from this
point of view everything really OK - are the calculated
Si charges usable ? Also PREPIN file contains info about bond
lengths, angles, dihedral angles but this are stricly related
to the actual values of the input molecular structure not the true
equilibrium ones am I right ?
Thanks a lot to anybody for any info or own experience with solving
of similar problem or just for reference
to relevant information resources (is there for example any
apprpriate tutorial available ?) !
Marek
####-AN---ORIGINAL---EMAIL
------- Pøedaná zpráva -------
Od: "Marek Malý" <maly_at_sci.ujep.cz>
Komu: amber_at_ambermd.org
Kopie:
Pøedmìt: [AMBER] Parametrisation of Silicon based molecules for Amber
simulations ?
Datum: Fri, 20 Mar 2009 00:49:21 +0100
Dear Amber users,
I would like to simulate molecules which contain also Si (silicon) atoms.
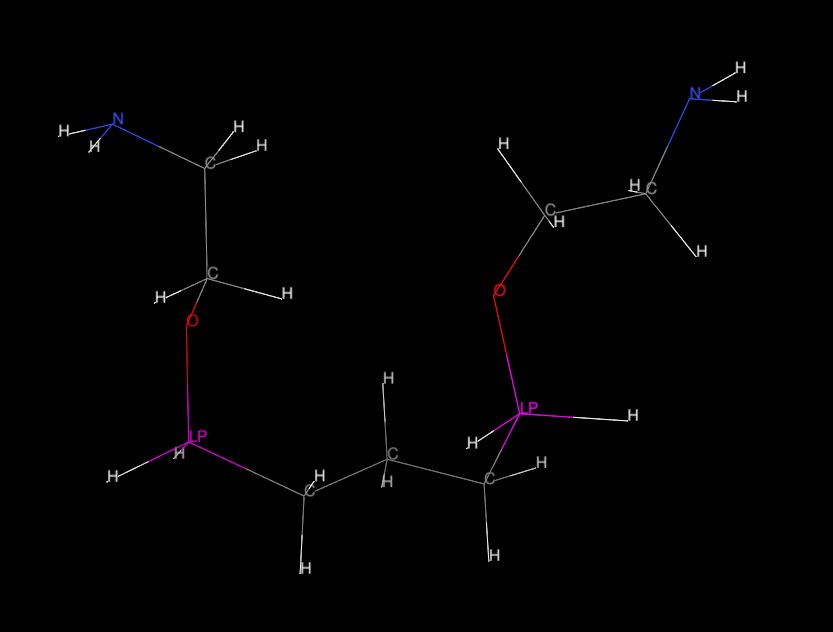
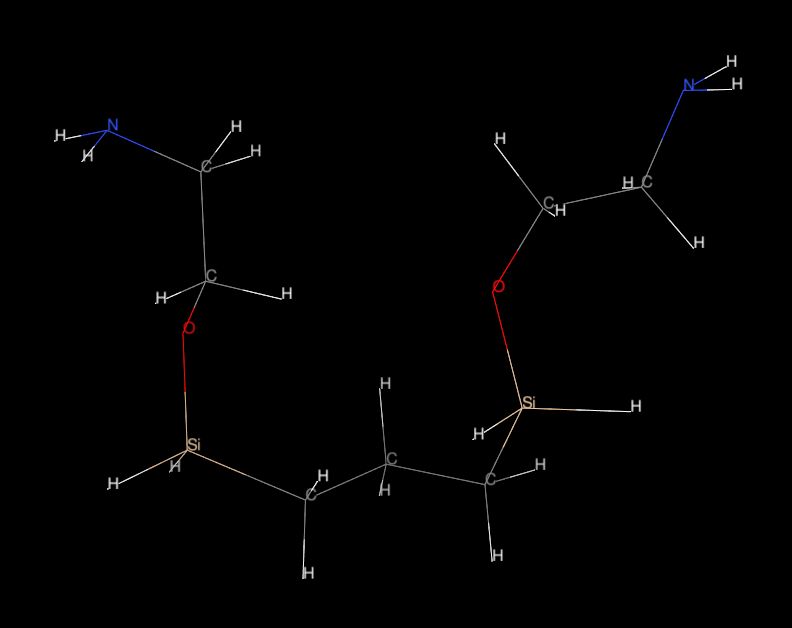
Please see attached fig. "mol01.jpg" or "mol.pdb."
I tried to parametrise small "testing" molecule using Antechamber and
I have obtained relevant PREPIN ("mol.prepin") and FRCMOD ("mol.frcmod")
files.
The prepin file is OK and also "Si" atoms were asigned with calculated
partial charge etc.
But there is problem with FRCMOD file which should be probably interpreted
in this case only as
an empty template for proper forcefield parameters (bond, angle, dihedrals
parameters) which
need to be included "manually".
Nevertheless I tried to make PRMTOP and INPCRD file (just for the
curiosity) and I succeeded,
but after reading of this two files by "Chimera" and labeling the element
types, the "Si" atoms
were asigned with the "LP" label (please see "mol02.jpg") although in
PRMTOP file are all "Si" atoms
named properly and in section "FLAG AMBER_ATOM_TYPE" is clearly written
type "Si". But this is probably
question for "Chimera" forum :))
It is clear to me that I will have to find a proper forcefield parameters
so
I am kindly asking for any info, links to relevant databases etc.
For example I succeed to simulate this molecule in vacuum with MS software
using "Dreiding" forcefield.
Could be a good idea to adopt desiderative parameters from this forcefield
and incorporate them into my FRCMOD
file ?
How to decide between for example two slightly different parametrisations
which I could find in different data sources ( articles, databases ) ?
Is there any highly recommended forcefield database available ?
I appreciate any help and advices especially from the experienced
researchers.
Thank you very much in advance !
Marek
-- Tato zpráva byla vytvoøena pøevratným po¹tovním klientem Opery: http://www.opera.com/mail/
- application/octet-stream attachment: mol.prepin
- application/octet-stream attachment: mol.inpcrd
- application/octet-stream attachment: mol.frcmod
- application/octet-stream attachment: mol.pdb
- application/octet-stream attachment: mol.prmtop
_______________________________________________
AMBER mailing list
AMBER_at_ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER_at_ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
- Previous message: Seth Hayik: "Re: [AMBER] make test.serial.QMMM"
- In reply to: Marek Malý: "[AMBER] Parametrisation of Silicon based molecules for Amber simulations ?"
- Next in thread: Karl Kirschner: "Re: [AMBER] FF Parameters Transformations ...?"
- Reply: Karl Kirschner: "Re: [AMBER] FF Parameters Transformations ...?"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
|
VU Home |
VUMC Home |
People Finder |
University Calendar
webmaster- modified on April 06, 2009 |