 | |||||||||||||||
 |
 |
 |
|
 |
|
 |
|
 |
|
 |
|
 |
|
 |
 |
AMBER Archive (2006)
Subject: AMBER: How to analyze the trajectory generated by LES simulation
From: pang zhao (zhao.pang_at_gmail.com)
Date: Fri Jun 02 2006 - 02:08:03 CDT
- Next message: Zhihong Yu: "AMBER: attachments for my last mail"
- Previous message: Zhihong Yu: "AMBER: some questions and problems about antechamber and divcon"
- Next in thread: Vlad Cojocaru: "Re: AMBER: How to analyze the trajectory generated by LES simulation"
- Reply: Vlad Cojocaru: "Re: AMBER: How to analyze the trajectory generated by LES simulation"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
Dear All,
I performe a LES simulation for 500 ps with 5 copys of the ligand.
I first run 2 ns NPT MD with explicit solvent and then with use the restrt
file "2ns.restrt" and prmtop file "npt.prmtop"
to generate the two files for LES, les.top, les.crd. I successfully run 500
ps LES simulation.
But I could not know how to analyze the trajectory generated by LES
simulation. Because the LES trajectory is different
from the normal trajectory.
when I try to do analyze hydrogen bond using ptraj with the file les.top,
the error occurs: ERROR in readParm: ...failed to find SOLVENT_POINTERS
When I use npt.prmtop for the ptraj analysis, it works.
But I find the analysis result is too different from that with the first 2
ns NPT trajectory.
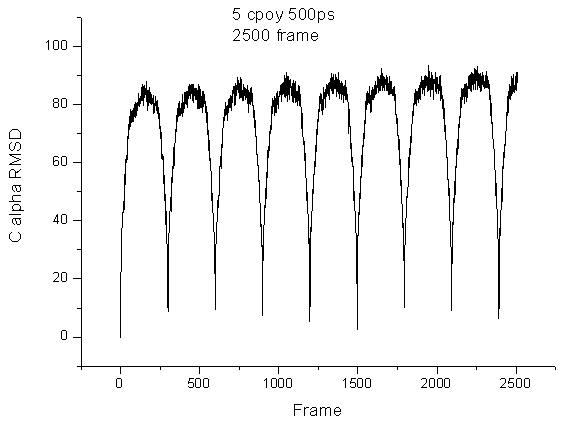
Such as the RMSD analysis for the LES trajectory, for the protein the C
alpha RMSD is too big.
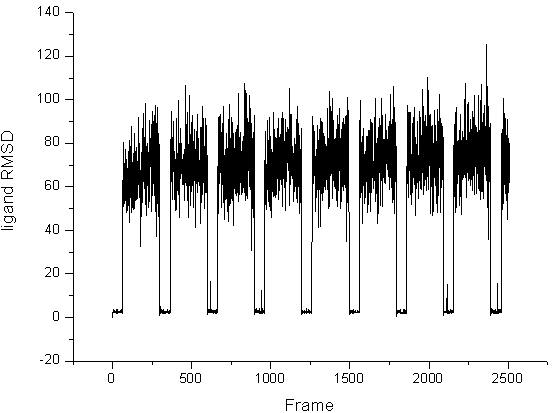
the picutre about the RMSD analysis is attached, one is the C alpha RMSD,
the second is that of the ligand.
the ptraj input :
trajin lesnpt.mdcrd
center :1-343 origin mass
image origin center familiar
rms first out protein_les_rms.out :1-342_at_CA
rms first out lig_les_rms.out :343
For the hydrogen analysis, I could not find the hydrogen bond of the ligand
with the LES trajectory,
but I can find the two hydrogen bonds of the ligand with the 2 ns NPT
trajectory.
Because the LES trajectory is different from the normal trajectory. I think
there the LES analysis may be diffrent from
the Normal MD simulations. But How to correctly analyze the LES trajectory?
Can I use the LES trajectory for MM-PBSA?
Great thanks!
 -----------------------------------------------------------------------
-----------------------------------------------------------------------
The AMBER Mail Reflector
To post, send mail to amber_at_scripps.edu
To unsubscribe, send "unsubscribe amber" to majordomo_at_scripps.edu
- Next message: Zhihong Yu: "AMBER: attachments for my last mail"
- Previous message: Zhihong Yu: "AMBER: some questions and problems about antechamber and divcon"
- Next in thread: Vlad Cojocaru: "Re: AMBER: How to analyze the trajectory generated by LES simulation"
- Reply: Vlad Cojocaru: "Re: AMBER: How to analyze the trajectory generated by LES simulation"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
|
VU Home |
VUMC Home |
People Finder |
University Calendar
webmaster- modified on January 30, 2009 |