 | |||||||||||||||
 |
 |
 |
|
 |
|
 |
|
 |
|
 |
|
 |
|
 |
 |
AMBER Archive (2005)
Subject: AMBER: TI - frcmod file
From: Oliver Hucke (ohucke_at_u.washington.edu)
Date: Tue Jul 12 2005 - 14:21:44 CDT
- Previous message: opitz_at_che.udel.edu: "Re: AMBER: MM_PBSA help, please"
- Next in thread: Ilyas Yildirim: "Re: AMBER: TI - frcmod file"
- Reply: Ilyas Yildirim: "Re: AMBER: TI - frcmod file"
- Reply: Douali, Latifa: "FW: AMBER: TI - frcmod file"
- Reply: Rhoad, Jonathan S.: "RE: AMBER: TI - frcmod file"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
Dear All,
I am setting up a TI simulation of the change of an ALA in a ligand
binding site of a protein to a GLY. The attached jpeg file shows the
changes of atom types I apply. In the process of preparing the frcmod
file for the atom type changes many errors of the following type
appeared when I tried to use my frcmod file with leap:
--- from leap output ---
*** Proper torsion parameters missing ***
atom names: CB-CA-C-N
atom types: CT-CT-C-N =pert=> H0-CT-C-N
Please add a dummy parameter of multiplicity 1
for the pert types to your parameter set.
- e.g. H0-CT-C-N 1 0.0 0. 1.
(This is because multiple torsional potentials may apply to a
single torsion, and each is perturbed individually in gibbs.)
--- end of excerpt from leap output ---
After doing what leap recommended, i.e. after adding all recommended
dummy torsions to my frcmod file, everything seems to be fine, i.e. I am
able to write the prmtop and crd output files with leap. (My frcmod file
that can be used with leap without problems is also attached).
BUT: some of the dummie torsions I had to add were actually "real"
torsions defined in the force field that I am using (ff03).
For example, leap asked me to add the following dummy torsion:
H0-CT-C-N 1 0.0 0. 1.
But in frcmod.03 I find:
H0-CT-C -N 1 0.0110 0.00 -2.
H0-CT-C -N 1 1.0607 180.00 1.
Does using the dummy torsion in my frcmod file for the perturbation not
change these parameters defined in frcmod.ff03?
Thank you very much,
Oliver
-- ________________________________________________________________Oliver Hucke, Dr. Biomolecular Structure Center Health Sciences Building - K418C Dept. of Biochemistry 1959 NE Pacific St. University of Washington phone: (206) 685 7046 Box 357742 fax : (206) 685 7002 Seattle, WA 98195-7742 email: ohucke_at_u.washington.edu ________________________________________________________________
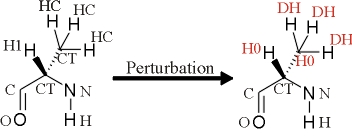
# Parameters needed for the perturbation of atom types
MASS
DH 1.008 0.135
BOND
H0-DH 340.0 1.090 CT-H0
H0-DH 340.0 1.090 CT-HC
ANGLE
CT-H0-DH 50.0 109.50 CT-CT-HC
C -CT-H0 63.0 111.10 C -CT-CT
N -CT-H0 80.0 109.70 CT-CT-N
DH-H0-DH 35.0 109.50 HC-CT-HC
DIHE
X -CT-H0-X 9 1.40 0.0 3. X -CT-CT-X
H0-CT-C-N 1 0.0 0. 1. dummy requested by leap; non-dummy in frcmod.ff03
H0-CT-C-N 1 0.0 0. 2. dummy requested by leap
H0-CT-C-N 1 0.0 0. 3. dummy requested by leap
H0-CT-C-O 1 0.0 0. 1. dummy requested by leap
H0-CT-C-O 1 0.0 0. 2. dummy requested by leap
H0-CT-C-O 1 0.0 0. 3. dummy requested by leap
H1-CT-C-N 1 0.0 0. 1. dummy requested by leap
H1-CT-C-N 1 0.0 0. 2. dummy requested by leap
H1-CT-C-N 1 0.0 0. 3. dummy requested by leap
H1-CT-C-O 1 0.0 0. 1. dummy requested by leap; non-dummy in parm99.dat
H1-CT-C-O 1 0.0 0. 2. dummy requested by leap
H1-CT-C-O 1 0.0 0. 3 dummy requested by leap
C-N-CT-H0 1 0.0 0. 1. dummy requested by leap; X -CT-N -X = dummy in parm99.dat
C-N-CT-H0 1 0.0 0. 2. dummy requested by leap
C-N-CT-H0 1 0.0 0. 3. dummy requested by leap
C-N-CT-H1 1 0.0 0. 1. dummy requested by leap
C-N-CT-H1 1 0.0 0. 3. dummy requested by leap
CT-CT-C-O 1 0.0 0. 1. dummy requested by leap; X -C -CT-X = dummy in parm99.dat
CT-CT-C-O 1 0.0 0. 2. dummy requested by leap
CT-CT-C-O 1 0.0 0. 3. dummy requested by leap
IMPR
NONBON
DH 1.0000 0.000
-----------------------------------------------------------------------
The AMBER Mail Reflector
To post, send mail to amber_at_scripps.edu
To unsubscribe, send "unsubscribe amber" to majordomo_at_scripps.edu
- Previous message: opitz_at_che.udel.edu: "Re: AMBER: MM_PBSA help, please"
- Next in thread: Ilyas Yildirim: "Re: AMBER: TI - frcmod file"
- Reply: Ilyas Yildirim: "Re: AMBER: TI - frcmod file"
- Reply: Douali, Latifa: "FW: AMBER: TI - frcmod file"
- Reply: Rhoad, Jonathan S.: "RE: AMBER: TI - frcmod file"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
|
VU Home |
VUMC Home |
People Finder |
University Calendar
webmaster- modified on January 30, 2009 |