 | |||||||||||||||
 |
 |
 |
|
 |
|
 |
|
 |
|
 |
|
 |
|
 |
 |
AMBER Archive (2005)
Subject: Re: AMBER: TI-FEP for ALA --> GLY
From: Jiten (jiten_at_postech.ac.kr)
Date: Mon Jun 27 2005 - 21:24:56 CDT
- Previous message: Ilyas Yildirim: "Re: AMBER: TI-FEP for VAL --> ALA"
- In reply to: Ilyas Yildirim: "Re: AMBER: TI-FEP for VAL --> ALA"
- Next in thread: Ilyas Yildirim: "Re: AMBER: TI-FEP for ALA --> GLY"
- Reply: Ilyas Yildirim: "Re: AMBER: TI-FEP for ALA --> GLY"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
Hai Ilyas Yildirim and amber users,
As I am using the FF03 with amber8 (with all_amino03.in, however I use xleap
of amber7 for making my input files) GLY has two H0 type attached to CT.
Therefore it would be fine with H0 atome type.
However, what is more concerning to me is on your comment
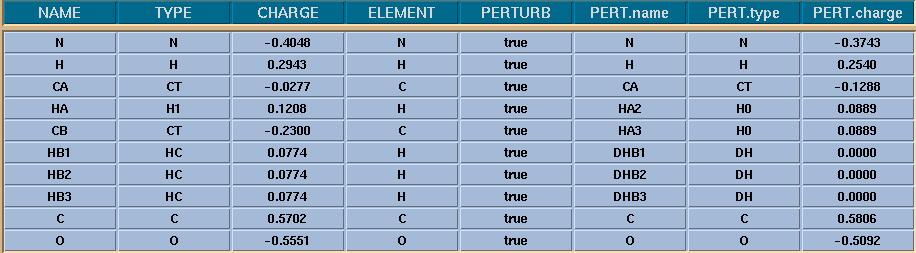
> I just realized something in your attached file: Your charges looks wrong.
> For instance, when u do the HC->DH transformation, the dummy atoms will
> have a zero charge in them. That means that the PERT.charge should be
> -0.0774 (according to AMBER 8). Which version of AMBER are u using? In
> Amber 8, the pert.charge is delta(charge), which is (charge of the state 1
> atom - charge of the state 0 atom). So, at state 1, u will have a dummy
> atom which has a zero charge in it. As a result (0 - 0.0774) = -0.0774
> should be in the PERT.charge column for this particular HC->DH
> transformation. You should do the same thing for the other atoms. If their
> final charge is different than the initial state charges, u should
> subtract them, and put that value into the PERT.charge column.
I am bit confused at this part. I have replaced the charges of ALA by GLY,
thereby the Dummy atom charges are set to zero. Therfore total pert.charge
of the system is also zero. Am I still doing wrong ?
Any suggestions will be highly appreciated.
Sincerely,
Jiten
----- Original Message -----
From: "Ilyas Yildirim" <yildirim_at_pas.rochester.edu>
To: <amber_at_scripps.edu>
Cc: "Jiten" <jiten_at_postech.ac.kr>
Sent: Tuesday, June 28, 2005 8:43 AM
Subject: Re: AMBER: TI-FEP for VAL --> ALA
> Hi Jiten,
>
> I just checked out the ALA and GLY residues. GLY residue has two H1 type
> hydrogen attached to CT type of carbon. So, I would do a CT->H1
> transformation, rather CT->H0 transformation. Generally, a CT type carbon
> has H1 type hydrogens, but in ALA residue, it has HC type hydrogen. Thats
> interesting. But in any case, I would do CT->H1.
>
> Good luck.
>
> On Tue, 28 Jun 2005, Jiten wrote:
>
>> Hi Ilyas Yildirim,
>>
>> In the system setup in solvent (second step of TI) for the TI method
>> using
>> xleap for A-->G, I have something like the attached file.
>>
>> My concern is CT --> HO : is it the right choice?
>>
>> Thanks for your help.
>>
>> Jiten
>>
>>
>> ----- Original Message -----
>> From: "Ilyas Yildirim" <yildirim_at_pas.rochester.edu>
>> To: <amber_at_scripps.edu>
>> Sent: Sunday, June 26, 2005 8:39 AM
>> Subject: Re: AMBER: TI-FEP for VAL --> ALA
>>
>>
>> > Hey Jiten,
>> >
>> > On Sun, 26 Jun 2005, Jiten wrote:
>> >
>> >> Hi Ilyas Yildirim,
>> >>
>> >> Thanks for the reply. First of all I apologize, what I have got zero
>> >> difference is of V--> A TI-FEP. The frcmod.DH was constructed as give
>> >> before
>> >> for ALA-->GLY.
>> >
>> > I assume by TI-FEP u mean free energy calculation using Thermodynamic
>> > Integration approach.
>> >
>> >>
>> >> First - I do not understand why I need use hydrogen mass for dummy
>> >> atom.
>> >> Second - I think that if I put the force constant and eq
>> >> distance/angle
>> >> for
>> >> the dummy atoms, with that just replaced by that off
>> >> !CT-HC, !HC-CT-HC,!HC-CT-HC etc -- I am confused if it still be
>> >> alright?
>> >>
>> >
>> > In the TI Approach tutorial, the dummy atoms had some mass. I think the
>> > reason is because of the mixed (hybrid) Hamiltonian. The forces are
>> > calculated according to this Hamiltonian. A zero mass might give an
>> > error,
>> > I think. The best choice is to use the mass of DH as
>> >
>> > MASS
>> > DH 1.008
>> >
>> > If it was a transformation from C->DC (dummy Carbon), it is better to
>> > use
>> > the mass of carbon for the mass of DC.
>> >
>> > For the second part of the question: I would do the same; defining the
>> > missing parameters by making some analogies with the old structure. In
>> > your case you have to define one more parameter, which is the dihedral
>> > angle of
>> >
>> > X -CT-HO-X
>> >
>> > Now, the reason why you have to have these force constants is to keep
>> > the
>> > structure in shape. The question is: Are these extra dummy atom
>> > parameters
>> > going to change the energy of the system? Yes they will but I do not
>> > think
>> > this is that crucial. These parameters are going to give some extra
>> > internal energy to the system, namely energy coming
>> > because of bonded parameters. In order to compare this free energy
>> > change
>> > with an experimental data, you have to do another free energy change
>> > simulation in gas phase (assuming you have done the first simulation in
>> > water). And then, u will subtract these 2 results. These
>> > internal/bonded
>> > energies will cancel at the end. So, that result will give you some
>> > reasonable prediction which can be compared with an experimental data.
>> >
>> > Here is what I mean:
>> >
>> > C-(H)3 ------------------> H-(DH)3
>> > (in water) G3 (in water)
>> > /\ /\
>> > || ||
>> > || ||
>> > || ||
>> > || G1 G2 ||
>> > || ||
>> > || ||
>> > || G4 ||
>> > C-(H)3 ------------------> H-(DH)3
>> > (in gas) (in gas)
>> >
>> > G1 and G2 are the experimental results, which are the solvation free
>> > energies. G3 and G4 are supposed to be the free energy simulation
>> > results.
>> > So,
>> >
>> > G1 - G2 = G3 - G4
>> >
>> > What I mean is, you have to do two free energy calculation, one for the
>> > solvated case, one in gas phase in order to compare it with an
>> > experimental result.
>> >
>> >> Also, I expect that the total energy after 12 windows be lower than
>> >> the
>> >> first window (similar to that of MM-PBSA), but they are not as given
>> >> below.
>> >> The structure after 12th window is still alright - nothing unusual.
>> >> Therefore, I feel that there something very wrong with it.
>> >>
>> >> Amber TI-FEP expert would help me ???
>> >>
>> >> MM-PBSA A V
>> >> ELE -8.44 -8.02
>> >> VDW -36.38 -36.02
>> >> GAS -44.82 -44.04
>> >> PBSUR -3.14 -3.01
>> >> PBCAL 23.04 24.59
>> >> PBSOL 19.91 21.58
>> >> PBELE 14.61 16.58
>> >> PBTOT -24.91 -22.46
>> >> TSTOT 18.48 17.58
>> >> PBTOT-S -6.43 -4.88
>> >>
>> >>
>> >> This is in good agreement with experiment.
>> >>
>> >> From first window,
>> >>
>> >> A V E R A G E S O V E R 50000 S T E P S
>> >>
>> >>
>> >> NSTEP = 100000 TIME(PS) = 120.000 TEMP(K) = 299.17 PRESS
>> >> =
>> >> 0.0
>> >> Etot = -77963.7518 EKtot = 19966.8759 EPtot
>> >> = -97930.6277
>> >> BOND = 722.3367 ANGLE = 1873.6537 DIHED =
>> >> 2212.5509
>> >> 1-4 NB = 935.9809 1-4 EEL = 11755.7341 VDWAALS =
>> >> 11246.8856
>> >> EELEC = -126677.7697 EHBOND = 0.0000 RESTRAINT =
>> >> 0.0000
>> >> DV/DL = 768.0586
>> >> Ewald error estimate: 0.6501E-04
>>
>> >> > ------------------------------------------------------------------------------From
>> >> 12th window, A V E R A G E S O V E R 50000 S T E P S NSTEP =
>> >> 100000 TIME(PS) = 1440.000 TEMP(K) = 299.59 PRESS =0.0 Etot
>> >> -77806.2969 EKtot = 19994.8561 EPtot = -97801.1530
>> >> BOND
>> >> = 712.4664 ANGLE = 1866.1103 DIHED =2207.5175 1-4
>> >> NB
>> >> = 933.9198 1-4 EEL = 11775.2910 VDWAALS =11186.6417
>> >> EELEC
>> >> = -126483.0998 EHBOND = 0.0000 RESTRAINT =0.0000 DV/DL
>> >> =
>> >> 0.0000 Ewald error estimate:
>> >> 0.6711E-04 ------------------------------------------------------------------------------The
>> >> dV/dl are as follows, 1 2 3 4 5 6 7 8 9 10 11 12 lam(i)
>> >> 0.00922 0.04794 0.11505 0.20634 0.31608 0.43738 0.562620.68392 0.79366
>> >> 0.88495 0.95206 0.99078 w(i) 0.02359 0.05347 0.08004 0.10158
>> >> 0.11675 0.12457 0.12457 0.116750.10158 0.08004 0.05347 0.02359
>> >> 768.05860 361.0522012.45120 -118.71170 -113.01570 -65.58740 -2
>> > 8!
>> >> .53030 -10.29490 -2.07680 -0.17480 -0.00800 0.00000 Total
>> >> 18.11850
>> >> 19.305460.99659 -12.05873 -13.19458 -8.17022 -3.55402 -1.20193 -0.21096
>> >> -0.01399
>> >> -0.00043 0.00000 0.01569Sincerely,Jiten----- Original
>> >> Message -----From:
>> >> "Ilyas Yildirim" <yildirim_at_pas.rochester.edu>To:
>> >> <amber_at_scripps.edu>Sent:
>> >> Saturday, June 25, 2005 3:48 PMSubject: Re: AMBER: TI-FEP for ALA -->
>> >> GLY> Hi Jiten,>> The frcmod file u created looks ok, but why do u have
>> >> 0
>> >> mass on the dummy> atoms? I would have chosen the hydrogen mass rather
>> >> than a zero mass for> the DH atom.>> Second thing, did u check out one
>> >> of
>> >> the trajectory file to see if> everything is ok? And also, what are
>> >> your
>> >> dv/dl results for these 12> lambda values. I have done a simple free
>> >> energy calculation for> ethane->methane which looks similar to what
>> >> you
>> >> have done (-CH3 changes to> H-DH3). I also got a result which was
>> >> close
>> >> to zero. I did the simulation> just for a test purpose. I have used
>> >> explicit solvent. In order to compare> with s
>> > o!
>> >> mething real (with an experimental data), u have to do 2 different>
>> >> TI
>> >> simulation: One in a solvent, the other in gas phase, and take the>
>> >> difference of these results. That will give u a prediction for an>
>> >> observable. (Thermodynamic Cycle)>> For the ethane->methane case, the
>> >> solvation of ethane and methane are> almost the same, so it is
>> >> reasonable
>> >> to expect something close to zero.> What is the solvation free
>> >> energies
>> >> of ALA and GLY (or the solvation free> energies of the initial and
>> >> final
>> >> states of your system)? The difference> of these energies should be
>> >> equal
>> >> to the free energy change of the> transformation in a solvent
>> >> subtracted
>> >> from the free energy change of the> transformation in gas phase.>>
>> >> Good
>> >> luck,>> On Sat, 25 Jun 2005, Jiten wrote:>>> Dear Amber users>>>> For
>> >> my
>> >> protein-drug system : while doing the free energy peturbationusing TI
>> >> method using 12 windows, and using the perl script attached here, Igot
>> >> almost zero difference in free energy. I used the following atom
>> >> types,where DH is dummy atom with zero charge and the charges of
>> > t!
>> >> he alaline isreplaced by glycine charges in the pert.charge
>> >> options>>>>>> H1 HC H0 DH>> |
>> >> |
>> >> | |>> --CT---CT--HC ---> --CT --H0--DH>> |
>> >> |>> HC DH>>>> My frcmod.DH is
>> >> as
>> >> follows,>>>> ALA to GLY>> MASS>> DH 0.000>>>> BOND>> H0-DH
>> >> 340.0 1.090 !CT-HC>>>> ANGLE>> DH - H0 - DH 35.0 109.50
>> >> !HC-CT-HC>> CT - H0 - DH 35.0 109.50 !HC-CT-HC>>>>
>> >> DIHE>> - The default values as given by leap>> -->> -->>>> NONB>> DH
>> >> 0.000 0.00>>>> Am I doing something stupid here ? Do I need to set
>> >> the
>> >> Bond and angles :K and R values as zero ?>>>> However, I calculate the
>> >> absolute free energy of the two syatems usingMM-PBSA from the last 1ns
>> >> trajectory of the 2ns MD simulations (wellconverged - rmsd, temp and
>> >> energy) and I got that there is clear differencesbetween the two
>> >> protein
>> >> systems.
>> > >!
>> >> >>> ELE -8.99 -8.96>> VDW -41.125 -37.68>>
>> >> GAS
>> >> -50.115 -46.64>> PBSUR -3.025 -3.07>> PBCAL 27.105
>> >> 24.16>> PBSOL 24.09 21.09>> PBELE 18.12 15.2>>
>> >> PBTOT -26.025 -25.55>> TSTOT 12.675 15.79>>
>> >> PBTOT-S -13.35 -9.76>>>>>> Thanks for any suggestions,>>>>
>> >> Sincerely,>>>> N. Jiten Singh>> C/O Prof. Kwang S. Kim>> Department of
>> >> Chemistry>> Pohang University of Science and Technology>> San 31,
>> >> Hyojadong, Namgu>> Pohang 790-784, Korea>> Phone : 82-54-279-5853 (
>> >> Lab )
>> >> / 279-4138 ( Appt )>> Fax : 82-54-279-8137 (or +82-54-279-3399)>> Web
>> >> :
>> >> http://csm.postech.ac.kr/ and http://www.postech.ac.kr/e>> Home Page :
>> >> http://www.geocities.com/njs_19>> --> Ilyas
>> >>
>> >> ldirim> --------------------------------------------------------------->
>> >> - Department of Chemisty - -> - University of
>> >> Rochester - -> - Hutchison Hall, # B10 - -> - Rochester,
>> >> NY
>> >> 14627-0216 - Ph.:(585) 275 67 66 (Office) -> -
>> >>
>> >> ttp://www.pas.rochester.edu/~yildirim/ -> ------------------------------------
>> > -!
>> >> -------------------------->> ----------------------------------------------------------------------->
>> >> The AMBER Mail Reflector> To post, send mail to amber_at_scripps.edu> To
>> >> unsubscribe, send "unsubscribe amber" to majordomo_at_scripps.edu>>>
>> >>
>> >
>> > I wrote a small script to calculate the free energy change of this
>> > simulation, and yes, it gave a deltaG = 0.15 (approx). But as I said,
>> > this
>> > result mean nothing. U have to do another simulation and subtract it
>> > with
>> > this one.
>> >
>> > If the only change is -CH3 --> -H, maybe it is reasonable. The
>> > experimental free energy of hydration of methane is 2.0 kcal/mol while
>> > it
>> > is 1.8 kcal/mol for ethane. The difference is 0.2 kcal/mol. (JACKS
>> > 1996, 118, 6285-6294; original data from Cabani et al. 1981) So, maybe
>> > the
>> > same thing is true for your system. Just a thought.
>> >
>> > Good luck,
>> >
>> >>
>> >>
>> >>
>> >> -----------------------------------------------------------------------
>> >> The AMBER Mail Reflector
>> >> To post, send mail to amber_at_scripps.edu
>> >> To unsubscribe, send "unsubscribe amber" to majordomo_at_scripps.edu
>> >>
>> >>
>> >
>> > --
>> > Ilyas Yildirim
>> > ---------------------------------------------------------------
>> > - Department of Chemisty - -
>> > - University of Rochester - -
>> > - Hutchison Hall, # B10 - -
>> > - Rochester, NY 14627-0216 - Ph.:(585) 275 67 66 (Office) -
>> > - http://www.pas.rochester.edu/~yildirim/ -
>> > ---------------------------------------------------------------
>> >
>> > -----------------------------------------------------------------------
>> > The AMBER Mail Reflector
>> > To post, send mail to amber_at_scripps.edu
>> > To unsubscribe, send "unsubscribe amber" to majordomo_at_scripps.edu
>> >
>> >
>> >
>>
>
> --
> Ilyas Yildirim
> ---------------------------------------------------------------
> - Department of Chemisty - -
> - University of Rochester - -
> - Hutchison Hall, # B10 - -
> - Rochester, NY 14627-0216 - Ph.:(585) 275 67 66 (Office) -
> - http://www.pas.rochester.edu/~yildirim/ -
> ---------------------------------------------------------------
>
>
> -----------------------------------------------------------------------
> The AMBER Mail Reflector
> To post, send mail to amber_at_scripps.edu
> To unsubscribe, send "unsubscribe amber" to majordomo_at_scripps.edu
>
>
>
-----------------------------------------------------------------------
The AMBER Mail Reflector
To post, send mail to amber_at_scripps.edu
To unsubscribe, send "unsubscribe amber" to majordomo_at_scripps.edu
- Previous message: Ilyas Yildirim: "Re: AMBER: TI-FEP for VAL --> ALA"
- In reply to: Ilyas Yildirim: "Re: AMBER: TI-FEP for VAL --> ALA"
- Next in thread: Ilyas Yildirim: "Re: AMBER: TI-FEP for ALA --> GLY"
- Reply: Ilyas Yildirim: "Re: AMBER: TI-FEP for ALA --> GLY"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
|
VU Home |
VUMC Home |
People Finder |
University Calendar
webmaster- modified on January 30, 2009 |