 | |||||||||||||||
 |
 |
 |
|
 |
|
 |
|
 |
|
 |
|
 |
|
 |
 |
AMBER Archive (2005)
Subject: Re: AMBER: Re:Re:single-stranded poly(rC) simulation
From: Thomas E. Cheatham, III (cheatham_at_chpc.utah.edu)
Date: Tue Apr 05 2005 - 13:32:15 CDT
- Next message: David A. Case: "Re: AMBER: doble C and N terminal"
- Previous message: Kateryna Miroshnychenko: "AMBER: Re:Re:single-stranded poly(rC) simulation"
- In reply to: Kateryna Miroshnychenko: "AMBER: Re:Re:single-stranded poly(rC) simulation"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
> Thank you for response. The problem is that the terminal conformation
> of my polyrC doesn't look like a helix much. May be you can recommend how
> to estimate the helicity of it? Is there any parameter ( or may be
> set of parameters) that could distinguish between "random coil" and
> distorted helical conformation?
You need some means to quantify your definition of "helical" vs. "coil".
As Professor Case mentioned, a 3 A RMSd is rather small. Also, RMSd is
not the ideal measure as a small rotation around the backbone (of the
terminal residue) could lead to a large RMSd.
In my experience with single stranded DNA homopolymers (10-mers),
structures started from a helical geometry (by deleting the second strand
of a B-DNA duplex) are metastable in a helical type conformation (by eye)
at least on a 5-15 ns time scale except for the terminal bases which (may)
show considerable motion (i.e. rotation around chi and blurring of the
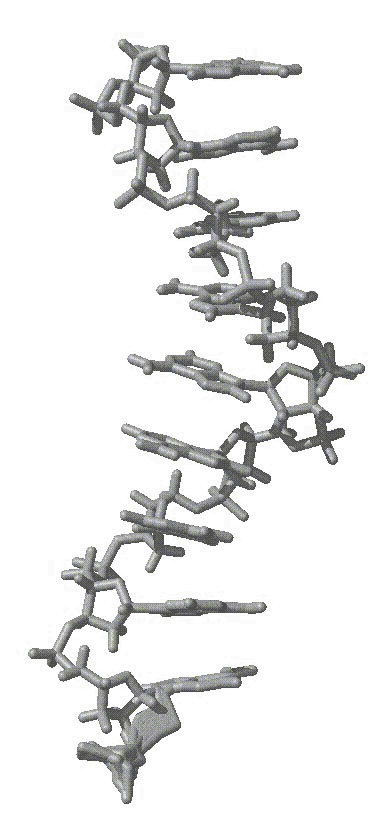
average structure as shown in the attached figure of an average structure
from initially helical polyA over 6 ns of MD, first attachement, gif).
If however, I (pseudo) randomly unfold polyA (via a method akin to high
temperature MD), over time the polyA tends to refold to a stacked geometry
from a completely unstacked geometry. This is not perfectly helical, but
"similar" and may include flipped out bases or loops. Folding RNA to a
helical geometry may be less likely (or harder or more time consuming) due
to the possibilities of intra-residue hydrogen bonding of the O2'
hydroxyl, the greater tolerance to mismatch structures, etc. However, to
my knowledge no one has looked at this yet in simulation (Nilsson's group
has published results on single stranded DNA; I have never published my
single strand work (dough!)).
If I were going to quantify single stranded nucleic acids:
(1) Backbone geometries (including sugar pucker) to characterize if angles
are within ranges expected for A-RNA [see Schneider et al. Biopoly 42,
113-124 (1997) for one set of "expected" backbone conformations]
(2) Helicoidal parameters per base pair step / base pair (twist, rise,
...) to see if these are within the expected range
(3) Some measure of "stacking" to see if continguous base stacking is
evident.
(4) Characterization of h-bonding within the structure (to see if it is
moving away from helical and self-interacting).
(5) Calculate a helical axis.
At present, there is no tool within AMBER to do helicoidal analysis or to
construct the helical axis, however there are many available programs out
in the ether ranging from Curves (Lavery group) to 3DNA (Olson/Lu) to
NEWHELIX (Dickerson) to ... Most of these work on snapshots (single
structures). The "Dials and Windows" interface to Curves (Wesleyan, $$$)
can look at trajectories, however it is used to handling double strands.
Note that different standards have existed for calculating these values
and there are differences whether one looks at "global" (with reference to
a helical axis) vs. "local" (base pair step) parameters. Papers by Olson
and co-workers provide a nice discussion of the issues.
ptraj can aid in looking at backbone angles (1) or h-bonding (4). There
is no automatic way to look at stacking, but one could use intra-base
distances (of center of mass) and/or a definition of a virtual dihedral to
somehow quantify stacking. One might also use an energy criteria (based
on non-bond attraction?).
Good luck.
--tom
\ Thomas E. Cheatham, III (Assistant Professor) College of Pharmacy, Depts of
| Medicinal Chemistry and of Pharmaceutics and Pharmaceutical Chemistry
| Adjunct Asst Prof of Bioengineering; Center for High Performance Computing
| University of Utah, 30 South 2000 East, Skaggs 201, Salt Lake City, UT 84112
|
| tec3_at_utah.edu (801) 587-9652; FAX: (801) 585-9119
\ BPRP295A / INSCC 418 http://www.chpc.utah.edu/~cheatham
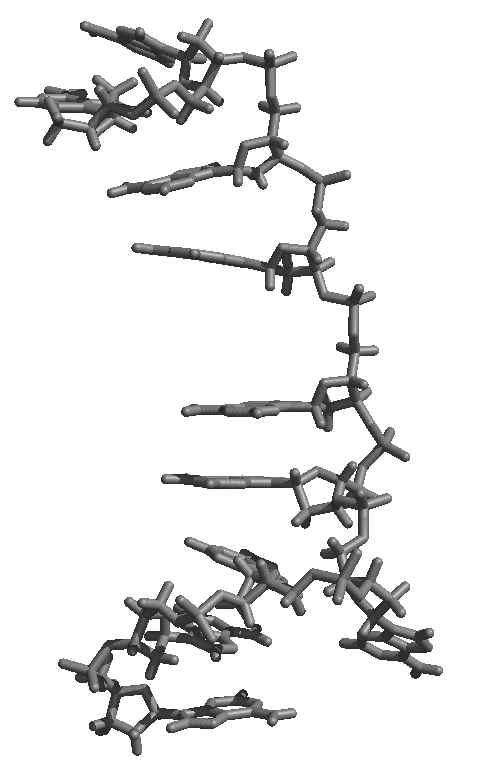 -----------------------------------------------------------------------
-----------------------------------------------------------------------
The AMBER Mail Reflector
To post, send mail to amber_at_scripps.edu
To unsubscribe, send "unsubscribe amber" to majordomo_at_scripps.edu
- Next message: David A. Case: "Re: AMBER: doble C and N terminal"
- Previous message: Kateryna Miroshnychenko: "AMBER: Re:Re:single-stranded poly(rC) simulation"
- In reply to: Kateryna Miroshnychenko: "AMBER: Re:Re:single-stranded poly(rC) simulation"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
|
VU Home |
VUMC Home |
People Finder |
University Calendar
webmaster- modified on January 30, 2009 |